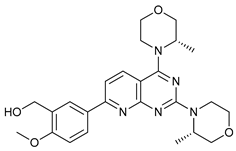
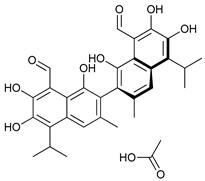
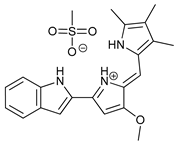
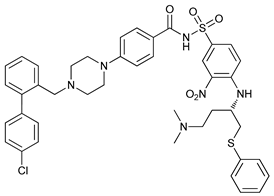
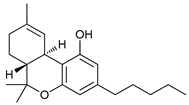
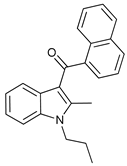
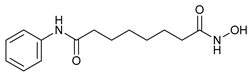
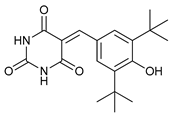
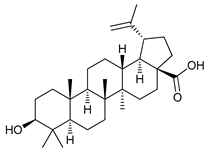
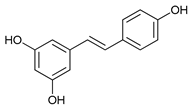
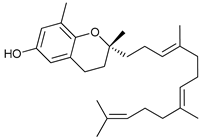
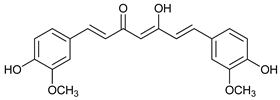
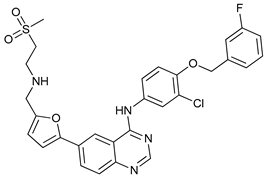
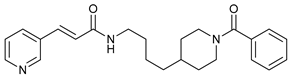
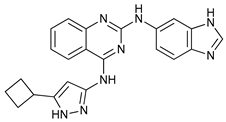
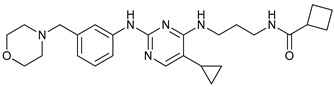
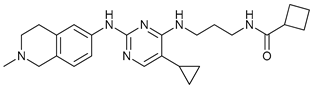
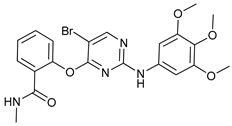
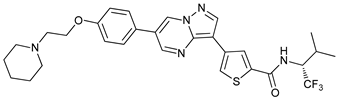
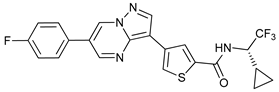
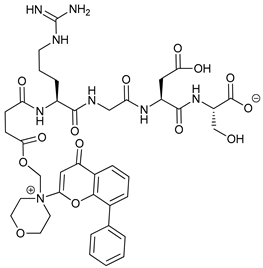
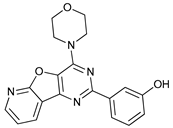
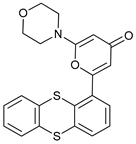
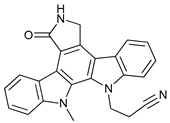
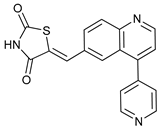
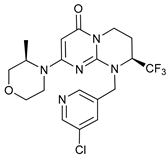
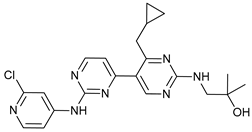
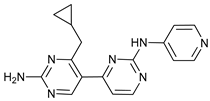
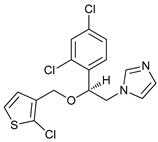
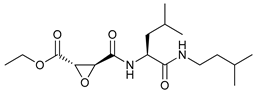
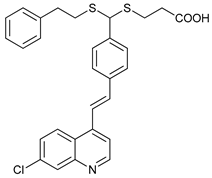
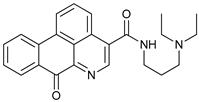
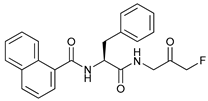
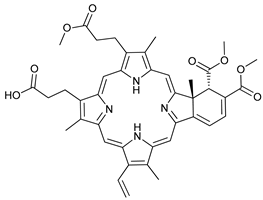
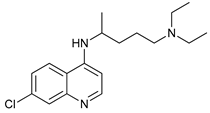
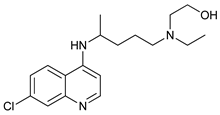
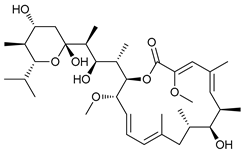
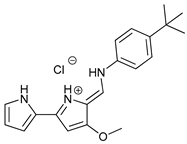
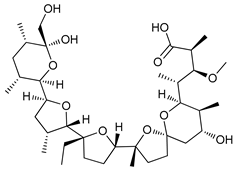
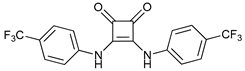
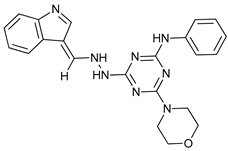
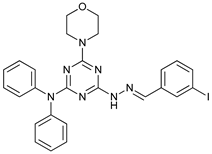
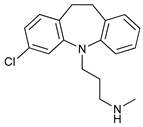
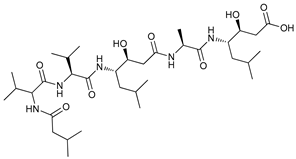
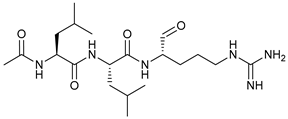
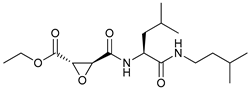
In several tumors, autophagy has a protective role; therefore, its inhibition could be an interesting approach for tumor treatment. There are several autophagy inhibitors that block the process of autophagy at different steps, which we detail below (Table 2).
2.2.1. ULK Inhibitors
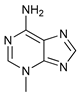
ULKs are a family of serine/threonine protein kinases that form complexes with multiple regulator units. The role of ULK1 is essential for the initiation of autophagy [
15,
160,
161], however the role of ULK2 in autophagy seems to be cell type dependent [
162]. Due to the homology between ULK1 and ULK2 [
163], inhibitors of ULK1 also inhibit ULK2 [
163]. ULK1 has been shown to be upregulated in several cancers, which correlated with poor prognosis and treatment resistance [
99,
164,
165,
166]. Inhibition of ULK1 has been shown to induce a decrease in tumor growth and induction of apoptosis [
100,
101]. This has led to the search for compounds that inhibit this kinase activity finding some molecules that compete with the ATP-binding site, such as compound 6 [
96], MRT68921, and MRT67307 [
97,
98]. Besides them, SBI-0206965 is the most studied [
102], which inhibits autophagy and induces apoptosis in neuroblastoma cell lines [
100], non-small cell lung cancer (NSCLC) cells [
101,
102], and in clear cell renal carcinoma cells [
99]. Moreover, it has also been reported to be a direct inhibitor of AMPK, which is a serine/threonine kinase that activates the ULK complex, among other roles [
167]. Recently more ULK inhibitors, such as ULK100 and ULK101, have been described [
103], which supports that the idea that blocking ULK1 may be a good strategy for cancer therapy.
2.2.2. Pan PI3K Inhibitors
The family of phosphoinositide 3-kinases (PI3Ks) is divided into three classes with different substrate preferences, which define their functions. The role of class II on autophagy is unclear. However, class I activates mTORC1 through the PI3K/Akt pathway and consequently inhibits autophagy, while class III (VPS34) activates autophagy [
168]. PI3K pathways have been associated with cancer due to their participation in tumorigenic processes such as cell proliferation, survival, migration, and angiogenesis. Therefore, they are a good target for therapy development [
169]. Most of the studied PI3K inhibitors are not selective for a specific class of PI3K, hence, they affect different cellular processes, not only autophagy, and consequently their effect cannot be only attributed to inhibition of autophagy. However, due to their therapeutic relevance, we describe briefly some of them below.
3-Methyladenine (3MA) was one of the first inhibitors of autophagy described [
104]. It exerts a dual effect on autophagy. Under starving conditions it suppresses autophagy through PI3KC3 inhibition. However, in the presence of nutrients it promotes autophagy by inhibition of PI3KC1 [
105]. Additionally, it has been reported that it reduces the expression of drug efflux transporters, overcoming taxol and doxorubicin resistance [
106]. 3MA is effective at high concentrations, although presents solubility problems. In order to overcome this limitation some derivatives have been synthetized [
107]. Wortmannin is a fungal metabolite that binds irreversibly to the catalytic site of PI3Ks [
108,
109]. LY294002 is a synthetic small molecule [
110] with poor solubility and short half-life. A conjugate analog of LY294002, named SF1126, was designed to accumulate in integrin expressing tissues, improving LY294002 solubility and pharmacokinetic, favoring its accumulation in the tumor site and showing antitumor and antiangiogenic properties in mouse models [
111,
112]. Other non-selective Pan PI3K inhibitors are PI103 [
113], KU55933, Gö6976 [
114], and GSK1059615 [
115,
116,
170].
2.2.3. VPS34 (PI3KC3) Complex Inhibitors
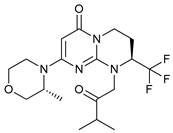
VPS34 is a PI3KC3 that transforms PI to PI3P. VPS34 forms a complex with several subunits needed for its activation, such as VPS15 (also known as p150), ATG14, and Beclin-1. Autophagy can be blocked by inhibition of VPS34 activity; SAR405 is one compound of the (2S)-tetrahydropyrimido-pyrimidinones series with kinase inhibitor activity by strong competition for ATP site. However, it is highly selective for PI3KC3, compared to class I and II, and more than 200 protein kinases and 15 lipid kinases. SAR405 inhibits autophagy induced either by starvation or mTOR inhibition [
113]. VPS34-IN1 is a bipyrimidinamine that inhibits PI3KC3 selectively, compared with more than 300 protein kinases analyzed [
117]. Additionally, PIK-III, a bisaminopyrimidine, binds to a hydrophobic pocket unique in VPS34 that cannot be found in other related kinases [
118]. Compound 31 is a small molecule selective against protein and other lipid kinases [
119]. All these four inhibitors are selective for PI3KC3, but it should be noted that VPS34 can form different complexes with other subunits that lead to a different localization and function, participating also in vesicle trafficking [
171]. Thus, inhibitors of VPS34 can also have an effect on endosomal trafficking, as the case of SAR405 that prevents the activity of both VPS34 complexes [
113]. Therefore, it may also affect cellular secretion [
172].
On the other hand, autophagy can be also inhibited blocking PI3KC3 complex formation; Spautin-1 indirectly inhibits the activity of VPS34 by proteosomal degradation of proteins that form VPS34 complexes through reduction of Beclin-1 deubiquitination mediated by USP10 and USP13 [
120].
2.2.4. ATG inhibitors
Membrane PI3P produced by VPS34 leads to the recruitment of PI3P-binding ATG proteins and additional factors, resulting in the formation of complexes that participate in the elongation of the phagophore. Inhibition of autophagy can be achieved by impeding the formation of these complexes.
ATG7 participates in the formation of the complex ATG12-ATG5 and the conjugation of PE to LC3 and GABARAP. Recently, some inhibitors of ATG7 (WO2018/089786) have been designed and it has extended the use of micro RNAs that target ATG7 gene such as miR-154 that inhibits blade cancer progression [
121].
On the other hand, ATG4B cleaves LC3, activating it for its conjugation with PE [
173] necessary for the expansion of the autophagosome and its recognition. Additionally, it participates in LC3-PE deconjugation, which is important for LC3 recycling and for the fusion of the autophagosome with the lysosome [
174]. Therefore, ATG4B could be a good target to inhibit autophagy more selectively, thus, a large number of ATG4B possible inhibitors have been screened in the last years [
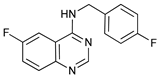
175]. NSC185058 is a small compound that docked at the active site of ATG4B inhibiting not only autophagy but also the volume of the autophagosomes, which is accompanied by suppression of tumor growth in an osteosarcoma subcutaneous mouse model [
122]. Tioconazole is an antifungal drug that binds to the active site of ATG4 blocking autophagy flux reducing cell viability and sensitizing tumor cells to doxorubicin in a xenograft mouse model [
123]. Other ATG4B inhibitors that suppress autophagy in cell lines and in vivo inhibiting cell proliferation are UAMC-2526, a derivative of benzotropolones stable in plasma [
124], and LV-320, a styrylquinoline [
125].
It should be noticed that the roles of ATG4B in cancer are not well understood and some of the ATG4 inhibitors showed only inhibition in LC3-PE delipidation, but not in the autophagosome formation such as S130 [
126] and FMK-9a [
127,
128,
129]. Additionally, some studies are focused on the evaluation of different markers that may predict the effectiveness of those inhibitors [
176]. For instance, ATG4B inhibition is effective only in Her-2 positive cells and not in those negative [
177].
2.2.6. Lysosome Inhibitors
The last step in autophagy is the fusion of autophagosomes with lysosomes, whose hydrolases degrade the autophagosome content. The inhibition of autophagy at this point consists of the use of lysosomal inhibitors.
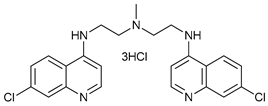
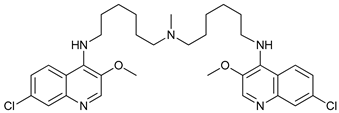
Chloroquine (CQ) and its analog hydroxychloroquine (HCQ) [
136] are drugs used for the treatment of various diseases, such as malaria and more recently cancer [
135]. They are weak bases and the unprotonated form of CQ/HCQ can diffuse through cell membranes and enter into organelles such as lysosomes, where the high concentration of H
+ induces their protonation and consequently increases lysosomal pH [
134]. Once CQ/HCQ are protonated, they are trapped in the lysosomes producing an increase of their volume, and inhibiting the activity of lysosomal enzymes.
CQ and HCQ are the only autophagy inhibitors approved for clinical use. Although short-term CQ/HCQ treatment has been considered safe, retinopathy has been reported produced by long-term treatment with HCQ in about 7.5% of patients [
182] and cardiotoxicity [
183]. The prevalence depends on the dosage and the duration of treatment [
184]. This toxicity limitation, along with inconsistencies in the results obtained in the clinic, have led to the study of new and more potent autophagy inhibitors [
185]. Thus, CQ analogs that exert more potent autophagy inhibitory activity have been synthetized. Lys05 is a dimeric analog of CQ that accumulates within acidic organelles, including lysosomes, more potently than HCQ [
137]. DQ661, a dimeric quinacrine (DQ), not only inhibits lysosomal catabolism, including autophagy, but also targets palmitoyl-protein thioesterase-1, resulting in the inhibition of mTORC1 signaling. DQ661 has shown effects on tumor mouse models alone and it also overcame resistance to gemcitabine [
139]. Another antimalaria compound found to inhibit autophagy with antitumoral properties is VATG-027 [
140]. On the other hand, mefloquine is also accumulated in lysosomes disrupting autophagy, it induces apoptosis and inhibits multidrug resistance protein1 (MDR1) being effective in multidrug-resistant tumor cells [
142]. Mefloquine sensitizes chronic myeloid leukemia (CML) cells derived from patients in chronic phase to TK inhibitors showing selectivity for stem/progenitor tumoral cells to normal cells [
141].
CQ and its derivatives are not the only drugs that target lysosomes to inhibit autophagy; GLP (ganoderma lucidum polysaccharide) is a polysaccharide from the fungus
Ganoderma lucidium with multiple antitumoral properties [
143]. GLP induces apoptosis in cancer cell lines [
145] and reduces tumor growth in mouse models [
144]. It has recently been seen that GLP impairs autophagy flux by reduction of lysosome acidification and the accumulation of autophagosomes has suggested to be the cause of apoptosis induction [
144]. Bafilomycin A (BafA) is a vacuolar-H
+ ATPase inhibitor that disrupts the acidification of lysosomes, vesicles, and vacuoles [
146,
147] by preventing the entry of H
+ into these organelles. BafA also inhibits the fusion of autophagosomes with lysosomes, by disruption of Ca
2+ gradients implied in this process [
148].
Ionophores can also disrupt lysosomal pH, impairing the autophagy process. Tambjamine analogues are anion selective ionophores derived from the naturally occurring tambjamines and induce mitochondrial swelling and autophagy blockade with cytotoxic effects in lung cancer cells and cancer stem cells (CSCs) [
149]. Monensin, nigericin, and lasalocid are cation ionophores, but only monensin presents selectivity for lysosomes [
150]. Squaramides are synthetic chloride transporters that also induce cell death by apoptosis [
151].
On the other hand, the WX8-family comprises five chemical analogs that disrupt the fusion of lysosomes with autophagosomes, lysosomes fission, and sequestration of molecules into the lysosomes without altering their pH. These compounds bind to PIKFYVE phosphoinositide kinase and present potent antitumoral effects on autophagic dependent cells [
152]. Vacuolin-1 activates RAB5A blocking the fusion of the autophagosomes with lysosomes, however it also inhibits the fusion of endosomes with lysosomes, resulting in a general endosomal-lysosomal degradation defective [
153].
Clomipramine (CM) is a FDA-approved prodrug for the treatment of psychiatric disorders the metabolite of which, desmethylclomipramine (DCMI), impairs autophagic flux blocking lysosomal degradation that sensitizes tumor cells to cancer treatment [
154]. DCMI also affects lung CSCs [
186]. Additionally, protease inhibitors can also inhibit the lysosomal degradation, such as pepstatin A (aspartyl proteases; cathepsin D and E), Leupeptin [
155] and E64d (cysteine proteases; cathepsin B, H, and L) [
156]. On the other hand, nanoparticles are usually accumulated into lysosomes by endocytosis internalization, which may cause lysosome impairment [
157]. Gold nanoparticles [
158] and nanodiamonds have shown to inhibit autophagy by disruption of lysosomal function, which sensitizes tumors to arsenical base therapy [
159].
Several studies have suggested that the anti-tumor effects of lysosomal inhibitors may be independent of autophagy inhibition since they also interfere in other cellular mechanisms producing non-autophagy related effects [
187,
188,
189,
190,
191,
192,
193,
194]. Remarkably, disruption of the lysosomes not only blocks autophagy, but lysosomal permeabilization releases proteases such as cathepsins that are active at cytosolic pH and participate in apoptosis and apoptosis-like and necrosis-like cell death [
195,
196,
197]. Additionally, lysosomes also participate in tumor invasion, hence, these inhibitors have shown to be effective against metastasis [
138,
198,
199,
200], targeting cancer stem cells [
201], and inducing tumor vessel normalization [
202].
As mentioned above, there are efforts to find genetic determinants to sensitivity or resistance to these lysosomal inhibitors. Metastatic cells are more vulnerable to CQ and BafA, suggesting that patients with metastasis could benefit from those treatments [
198]. Morgan and coworkers also showed a relationship between the expression of ID4 and metastatic potential. Additionally, overexpression of helicase-like transcription factor (HLTF) seems to be related with the resistance to HCQ, Lys05 and BafA treatment [
189] and tumors with the V600E mutation in BRAF (v-Raf murine sarcoma viral oncogene homolog B) present cytoprotective autophagy [
203].