Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
STAT3 is an important transcription factor that regulates cell growth and proliferation by regulating gene transcription of a plethora of genes.
- STAT3
1. Introduction
The signal transducer and activator of transcription (STAT) family is composed of seven members: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. These proteins work mainly as transcription factors that bind specific consensus palindromic sequences on DNA and share the same structure. STAT proteins are composed of the following: an N-terminal domain, which is responsible for dimerization, nuclear import, and tetramerization on target gene promoters with tandem binding sites; a coiled-coil domain containing the nuclear localization sequence (NLS) and consequently involved in nuclear import and export; a DNA binding domain (DBD) that binds promoters of target genes in consensus sequences; a linker region; a highly conserved SH2 domain involved in recognition of receptor subunits and in STATs dimerization by stabilization of STAT dimers; and a transactivation domain that mediates transcription of STAT target genes (Figure 1) [1,2]. The importance of these transcription factors in the control of cellular processes is known worldwide and has been confirmed by the strong conservation of Stat sequences in different eukaryotic organisms, from Hydra magnipapillata to Caenorabditis elegans, from Dictyostelium discoideum to Drosophila melanogaster, from Danio rerio to Homo sapiens [3]. Although all the STAT proteins have a pivotal role in cellular homeostasis, nowadays, the study of STAT3 has gained more importance because several functions of this protein have not yet been completely described and because it exerts many different functions in tumor growth and progression [4]. The canonical activity of STAT3 is triggered by the IL-6/JAK/STAT3 pathway which is induced after binding of interleukin (IL) 6 family member proteins (including IL-6, IL-11, ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), oncostatin M (OSM), cardiotrophin 1 (CT1), cardiotrophin-like cytokine (CLC), and IL-27) with membrane receptors, which are tethered to GP130 transmembrane proteins. GP130s physically interact with Janus kinases (JAKs) in their cytosolic portion and the interaction between IL-6 family members with their cognate receptors determines conformational changes to the autophosphorylation of JAKs. Phosphorylated JAKs trigger the phosphorylation of tyrosine 705 (Y705) of STAT3 protein and pSTAT3 Y705 dimerizes and migrates to the nucleus, where it interacts with its responsive elements on DNA (STAT3 inducible elements, SIEs), inducing the transcription of target genes (Figure 2) [5]. Alternatively, STAT3 can also be phosphorylated in Y705 residue after binding of growth factors such as epithelial growth factor (EGF), transforming growth factor α (TGFα), platelet-derived growth factor (PDGF) and hepatocyte growth factor (HGF) with their transmembrane receptors [6,7]. STAT3 forms homodimers, however, under specific conditions it can also interact with STAT1, STAT5a, STAT5b, and STAT4, exerting both inflammatory and anti-inflammatory functions [8]. STAT3 is involved in many biological processes such as stem cell pluripotency maintenance by regulating DNA methylation [9]; wound healing and regeneration [10]; inflammation [11,12]; and metabolism [13]. Of note, the role of this protein in tumor progression has been extensively studied and several types of cancers are characterized by high levels of STAT3 protein and its persistent activation [4,14,15]. However, it is not yet clear whether these functions of STAT3 rely on its nuclear activities or are also controlled by its localization in other subcellular compartments. STAT3 has been detected in mitochondria and endoplasmic reticulum where it regulates mitochondrial DNA transcription, electron transport chain activity, and calcium homeostasis [16,17,18,19]; however, neither its mechanism(s) of translocation nor the mechanisms of action of the transcription factor in these organelles have, so far, been completely understood. Similar to other STATs, STAT3 undergoes several post-translational modifications (PTM) which regulate the different functions of this protein in the nucleus as well as in other subcellular compartments [20,21]. With this review, we sum up the discoveries about phosphorylation, acetylation, and methylation, highlighting the discrepancies among several studies and the importance of targeting these PTMs to therapeutically target STAT3 in cancer.
2. Phosphorylation
STAT3 undergoes several PTMs and the most studied is phosphorylation, consisting of the covalent addition of a phosphate group to specific amino acids. This PTM drastically modifies the properties and functions of protein. The canonical IL-6/JAK/STAT3 pathway relies on the phosphorylation of STAT3 in Y705 residue in the carboxy-terminus side of the chain, catalyzed by activated JAKs after cytokine stimulation. This modification leads to STAT3 dimerization and nuclear translocation, allowing STAT3 to act as a transcription factor. In the nucleus, STAT3 dimers interact with DNA on the SIEs and regulate the transcription of genes involved in stem cell pluripotency maintenance (Klf4), regeneration and wound healing (Il10), inflammation (Il8rb and Cxcl2), apoptosis (caspase-3), and many other important biological processes (Figure 2) [5,22,23,24,25].
Another phosphorylation site of STAT3 is the serine 727 (S727) residue. The mechanisms triggering this modification and the function of phosphorylated S727 are still debated. The kinases that are able to induce this modification are cell and stimuli specific [26] and they are mainly extracellular signal-regulated kinase (ERK) 1, ERK2, mitogen-activated protein kinase (MAPK) p38, c-Jun N-terminal kinase (JNK), and an H-7-sensitive kinase [27]. S727 phosphorylation has often been considered to be an enhancer of STAT3 nuclear transcriptional activities that probably acts by recruiting activating cofactors [27], such as NcoA [28] and CBP/p300 [29].
After the experiments of Boulton et al. [30] and of Zhang et al. [31], who discovered the STAT3 S727 phosphorylation on SDS-gel electrophoresis, Wen and collaborators [32] studied the functions of this PTM. They used COS monkey cells—characterized by low levels of Stat3 transcript—and overexpressed a mutated form of Stat3 in which S727 had been replaced with an Alanine, hence, blocking the possibility of STAT3 being phosphorylated in the 727 position (Stat3 S727A) (Figure 3). Interestingly, an electrophoretic mobility shift assay (EMSA), used to determine the capability of protein to bind DNA, demonstrated that the S727A mutation did not affect the ability of STAT3 to bind SIE. Finally, the authors used U3A cells (that showed a lower response of endogenous STAT3 to IFN-α stimulation, but a higher response when new Stat3 is introduced [33]) expressing Stat3 S727A to test its transcriptional activity with luciferase assay; the results confirmed that S727 phosphorylation was fundamental for maximal activation of STAT3 protein [32].
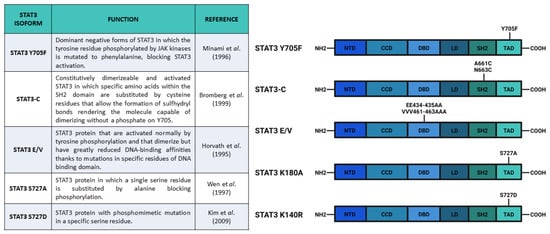
Figure 3. Main constructs used to investigate STAT3 phosphorylation. Created with BioRender.com.
In 2000, Decker and Kovarik [34] noticed some contrasting results reviewing information available about the function of S727 phosphorylation, while Schuringa et al. [35] confirmed in hepatoma cells stimulated with IL-6s the results of Wen et al., and Kim and Baumann [36] reported that STAT3 wild type and STAT3 S727A activities on hepatoglobin acute phase promoter were similar. Decker and Kovarik concluded that the function of S727 phosphorylation depended on the specific target gene promoter and/or cellular context. In addition, Decker and Kovarik reviewed the studies about the interdependence of serine and tyrosine phosphorylations. Considering that, in cells transfected with Stat3 S727A, there is upregulation of Y705 phosphorylation [37], but also taking into consideration the possible role of S727 phosphorylation in enhancing STAT3-dependent transcription [32,35], they proposed that S727 phosphorylation negatively affected Y705 phosphorylation as a direct effect, but also activated mechanisms (probably cofactor recruitment) that could overcompensate the negative effect on Y705 phosphorylation. From the functional point of view, Decker and Kovarik [34] supposed that the phosphorylation of S727 was important for STAT3-dependent control of cellular growth and they reported that overexpressed STAT3 S727A had a dominant-negative effect on transformation mediated by v-Src [38], which was also reduced after inhibition of serine phosphorylation of STAT3 protein [39].
Further analyses on STAT3 S727 functions were carried out in vivo by Shen et al. [40], who generated mice with the S727A substitution in the Stat3 gene (called SA allele). They used embryonic fibroblasts from homozygous Stat3SA/SA mice, and they discovered a halved transcriptional response in mutants as compared with wild type fibroblasts, hence, confirming the results described in Wen et al., [32]. Since Stat3SA/SA and Stat3+/- mice did not show altered phenotypes, Shen and collaborators bred these two lines to generate Stat3SA/- mice. This composite heterozygous line showed perinatal lethality (about 75% of the offspring died for no specified reasons) and lower embryonic and perinatal growth. The authors proposed a connection between this early phenotype and the altered levels of insulin-like growth factor (IGF-1) in the serum of newborn mice. According to the aforementioned data, fibroblasts from Stat3SA/- foetuses showed no more than 25% of STAT3-dependent transcription after IL-6 stimulation. Finally, Stat3SA/- mice showed a decreased level of thymocytes and an increased apoptosis, suggesting the presence of defects in the thymocyte survival mechanisms. Although this study did not dissect the functions of S727 phosphorylation in different tissues and the authors analyzed only fibroblasts from foetuses, the Stat3SA/SA murine line represented a good tool to further investigate STAT3 S727 phosphorylation in an in vivo system.
More recently, Hazan-Halevy et al. [41] also discovered a cellular model to investigate the role of S727 phosphorylation. Although STAT3 Y705 is frequently constitutively phosphorylated in solid and hematologic tumors, they showed that in chronic lymphocytic leukemia (CLL), which is the most common leukemia in the Western hemisphere, STAT3 is constitutively phosphorylated in S727 residue rather than in Y705, which in turn can be transiently phosphorylated in this cellular model. In detail, Hazan-Halevy and collaborators demonstrated that S727 residue of STAT3 was constitutively phosphorylated in the peripheral leukemic blood cells (CD19+) from patients with CLL as compared with non-leukemic (CD19-) cells from the same patients and healthy donors. The authors also demonstrated that pSTAT3 S727 translocated into the nucleus and bound to DNA, regulating STAT3-target genes. pSTAT3 S727 was found in nuclear fractions of CLL cells and was also detected by confocal microscopy. In the nuclei of unstimulated CLL cells, pSTAT3 S727 interacted with SIEs, as revealed by EMSA and ChIP assays and activated the transcription of specific STAT3-dependent genes. According to these results, the binding of STAT3 to DNA was not affected by dephosphorylation in Y705 residue. Finally, they concluded that pSTAT3 S727 could be a specific marker and a therapeutic target for CLL [41].
As previously mentioned, phosphorylation of both Y705 and S727 has been widely correlated to cellular growth control and cancer onset. For example, in these years, it was demonstrated that progestins were able to activate STAT3 in breast cancers by inducing phosphorylation of Y705 or S727 [26,42]. Proietti and collaborators [42] showed that in murine (C4HD) and human (T-47D) breast cancer cells, treatment with the synthetic progestin medroxyprogesterone acetate (MPA) promoted phosphorylation of Y705 STAT3 and consequent nuclear translocation, SIE binding, and STAT3-dependent transcriptional activation mediated by JAK- and Src-dependent pathways. According to their results, the MPA treatment stimulated phosphorylation of STAT3 (Y705), JAK1, JAK2, and Src, and this effect was abolished by the progestin antagonist RU486, indicating the direct involvement of progestin receptor (PR) (demonstrated to be an interactor of STAT3 upon MPA stimulation). Notably, the phosphorylation of STAT3 on Y705 was completely abrogated after inhibition of JAK1, JAK2, and Src. Moreover, the authors demonstrated a correlation between progestin stimulation of breast cancer growth and progestin induction of pSTAT3 Y705 and its subsequent transcriptional activity. By transfecting C4HD cells with Stat3C and Stat3 Y705F (acting, respectively, as dominant active and dominant negative forms of Stat3) (Figure 3), Proietti and collaborators demonstrated that the MPA-stimulated growth depended on STAT3 phosphorylation and its consequent activity. The cells transfected with Stat3 Y705F showed lower growth rates and higher levels of apoptosis, suggesting that the dominant negative form of STAT3 determines growth inhibition by cell cycle arrest and apoptosis. In order to verify this effect in vivo, Proietti and collaborators inoculated the transfected cells in mice treated with MPA depot. Only a few mice injected with cells expressing Stat3 Y705F developed tumors and these tumors had a lower growth rate, confirming that targeting STAT3 (and phosphorylation of its residues) could be a very effective therapeutic strategy against breast cancer.
According to Tkach et al. [26], treatment with the synthetic progestin MPA could also induce S727 STAT3 phosphorylation in murine (C4HD) and human (T-47D) breast cancer cells and this induction was mediated by the PR. The effect was abolished by progestin antagonist and PR gene knockdown (by siRNA) in C4HD cells or by stable PR knockout in T-47D cells (in which the inducibility was restored upon PR transfection). The authors hypothesized that S727 phosphorylation was induced by progestins through activation of the c-Src/p42/p44 MAPK pathway, inhibition of which (using either specific inhibitor or genetic mutations) blocks S727 phosphorylation after MPA stimulation. Tkach et al. also showed that phosphorylation of S727 increased STAT3 nuclear translocation and maximized STAT3 transcriptional activities. In detail, the S727 phosphorylation of STAT3 was fundamental for the full transcriptional activation of cyclinD1 gene (a key cell cycle regulator in breast cancer) through pSTAT3 S727 recruitment on its promoter. Finally, they demonstrated that phosphorylation of S727 was needed for progestin-induced tumor growth, both in vitro and in vivo (through the injection of C4HD cells in BALB/c mice). The proliferation of the cells was reduced by transfection of Stat3 S727A vector in both murine (C4HD) and human (T-47D) breast cancer cell as compared with the control cells transfected with wild type Stat3 or empty vector. Notably, mice also injected with C4HD cells transfected with Stat3 S727A vector showed a reduced growth of tumor, in which the levels of pSTAT3 S727 and of cyclinD1 mRNA were lower as compared with the control [26].
In order to understand the role of STAT3 Y705 and S727 phosphorylation, Huang et al. [43] analyzed the different functions of these two PTMs in the fate choice of mouse embryonic stem cells (ESCs). These authors optimized an inducible system of STAT3 expression in Stat3-/- mESCs that could express either wild type Stat3, Stat3 Y705F, or Stat3 S727A. After inducing the different Stat3 forms and LIF stimulation, they showed that Stat3 expression was needed for cell survival and that Y705 phosphorylation was indispensable for the maintenance of pluripotency. In addition, their results showed that S727 phosphorylation was involved in cell survival and mitogenicity; cells expressing Stat3 S727A had reduced survival and proliferation rate as compared with cells expressing wild type Stat3. Moreover, also in mESCs, S727 was phosphorylated by ERK1/2 kinases. The role of S727 phosphorylation in the regulation of proliferation was probably connected to STAT3-dependent expression of Myc gene, which was abolished in mESCs expressing STAT3 Y705F and reduced in mESCs expressing STAT3 S727A. In addition, three other STAT3 target genes involved in the maintenance of pluripotency (Socs2, Nanog, and Klf4) had the same responsiveness of Myc, confirming the role of S727 phosphorylation as a transcription enhancer. The S727 phosphorylation also appeared to be involved in neuronal differentiation. Loss of pS727 resulted in a reduction in neuronal differentiation potential, recovered by the S727 phosphomimetic mutations, called Stat3 S727D, which substituted the serine with aspartic acid (Figure 3). Huang and coworkers also proposed an antagonistic role between S727 and Y705 phosphorylation in the reprogramming of epiblast-derived stem cells (EpiSC). The epiblast stem cell stage is an obligate transitional step for the mESC differentiation. During this stage, mESCs can be reprogrammed to naïve pluripotent stem cells by overexpression of STAT3. Huang and collaborators demonstrated that pSTAT3 S727 negatively affected this phenomenon. All in all, these authors suggested that a dynamic equilibrium between pS727 and pY705 determined fate decisions in mECSs through two distinct mechanisms, i.e., while Y705 phosphorylation is fundamental for self-renewal and pluripotency maintenance, S727 phosphorylation is involved in cell proliferation, survival, and pluripotency potential [43].
Specific interactors of STAT3 can affect its phosphorylation, altering the activities of the protein and leading to cell transformation. For example, in a study by Aziz et al. [44], they demonstrated that protein kinase Cε (PKCε) could interact with STAT3 in different types of human cancers. This protein has been previously defined as a transforming oncogene and a predictive biomarker for some types of human cancer. The STAT3-PKCε reciprocal immunoprecipitation has indicated this molecular interaction in skin melanomas, prostate, gliomas, bladder, colon, lung, pancreatic, and breast cancer cells [44,45,46,47]. Of note, the use of blocking peptide in the immunoprecipitation experiment inhibited this interaction, confirming the physical relation between PKCε and STAT3 suggested by Aziz and collaborators. According to their results, in some tumors (melanoma, glioma, pancreatic, and lung cancer cells) PKCε could induce phosphorylation of S727 residue leading to an increase in STAT3 transcriptional activity. The inhibition of PKCε by siRNA hampered, in turn, S727 phosphorylation (without affecting Y705 phosphorylation), STAT3 interaction with DNA, and STAT3-dependent gene expression. The effect of this inhibition resulted in a reduced invasiveness ability of cancer cells. PKCε silencing also reduced the activation of MAPK cascade, the inhibition of which further reduced PKCε-mediated S727 phosphorylation of STAT3. These data suggest that PKCε can mediate STAT3 S727 phosphorylation via MAPK cascade (RAF-1, MEK1/2, and ERK1/2) and that this mechanism is fundamental for the constitutive activation of STAT3 in human cancers mentioned above. Finally, the authors concluded that PKCε was an initial signal that induced STAT3 to sustain cancer invasiveness.
The state of STAT3 is also regulated by the protein tyrosine phosphatases (PTPs), which can dephosphorylate STAT3 residues. The mechanisms of PTP functions have only been partially clarified and are currently under investigation as possible targets for treatments against cancer [48].
The central role of STAT3 in the development of a large number of tumors makes this protein an attractive target for studies of cancer therapy. Starting from the observation that activated STAT3 is detected in a large fraction of lymphoid malignancies, Kuusanmaki et al. [49] looked for drugs that could inhibit WT STAT3, and also STAT3 isoforms with gain of function mutations (Y640F and D661V) leading to increased Y705 phosphorylation, as well as STAT3 dimerization and activation. They tested the drugs that could target STAT3 activity using different cell model systems (Ba/F3 cells, NK cell leukemia/lymphoma cells, and LGL leukemia patient samples) and identified four classes of drugs, among 306 approved compounds, that seemed to be effective against wild type STAT3 and its mutated isoforms, i.e., mTOR, JAK, Hsp90 and CDK inhibitors. After testing the different drugs in vivo, these authors concluded that JAK inhibitors could be an efficient in vivo therapeutic strategy because, even if they are less effective in cells expressing mutated forms of STAT3, they could inhibit microenvironmental cytokine stimulation and STAT3 hyperactivation even in STAT3-mutated malignancies. Nonetheless, the more promising treatments seemed to be Hsp90 inhibitors that exerted antitumoral functions both in cells expressing Stat3 WT, Stat3 Y640F, or Stat3 D661V. Finally, Kuusanmaki and collaborators concluded that Hsp90 inhibitors, also in combination with JAK or mTOR inhibitors, may be a very potent long-term therapeutic option for lymphoproliferative diseases characterized by STAT3 mutations.
After underlining the roles of both pY705 and pS727 of STAT3 in the regulation of nuclear activities, it is worthwhile mentioning that STAT3 can localize in different subcellular compartments, and exert functions that are independent from canonical STAT3 transcriptional activities [50,51,52]. Although the localization of STAT3 in mitochondria is still under investigation and its putative mechanism of mitochondrial translocation has not yet been described, Peron and collaborators [18] dissected the roles of Y705 and S727 phosphorylation in the mitochondrial activities of STAT3. In particular, using mESCs and zebrafish as an in vivo model, the authors demonstrated that STAT3 affected mitochondrial transcription and cell proliferation in mESCs and also in a specific pool of adult stem cells of zebrafish optic tectum. Using in situ hybridization, Peron and coworkers demonstrated that overexpression on murine Stat3 mRNA in zebrafish determined an upregulation of both mt_nd2 (used as a hallmark of mitochondrial transcription) and pcna (used as a marker of cell proliferation). Notably, the overexpression of Stat3 Y705F and Stat3 S727A did not stimulate expression of mt_nd2 and pcna, suggesting that phosphorylation of both Y705 and S727 was required for increased transcription of these genes. Hence, the authors decided to analyze the effects of a mitochondrially targeted form of Stat3 (named MLS_Stat3_NES) and demonstrated that phosphorylation of both Y705 and S727 was required for the correct activities of STAT3 in the mitochondrion. While Y705 appeared to be necessary for the proper localization of STAT3 in mitochondria, S727 was needed for the proper biological activities of this protein in the organelle.
In summary, Y705 phosphorylation is involved in the STAT3 canonical activation mechanism leading to dimerization and nuclear translocation of protein [5] but this PTM is also necessary for STAT3 mitochondrial localization [18]. This modification is largely correlated to STAT3 function as a nuclear transcription factor regulating cell cycle, cell pluripotency, and cell proliferation [43]. As reviewed in Avalle et al. (2019), alterations in Y705 phosphorylation have been related to cancer onset and progression [42] and, consequently, pharmacological treatments that are able to target Y705 phosphorylation, for example, Hsp90 inhibitors [40], are considered to be promising for cancer treatment [49]. Regarding S727 phosphorylation, the literature does not allow one to completely clarify its biological role. According to the reported information, S727 phosphorylation is not involved in STAT3 DNA binding [32], but it is necessary for nuclear translocation [26] and maximal activation of transcription (evidence confirmed in vitro by Wen et al. [32] and in vivo by Shen et al. [40]) in specific cellular and promoter contexts [34] probably through recruitment of cofactors useful for transcriptional activation [28,29]. S727 phosphorylation is also fundamental for the biological function of STAT3 in mitochondria [18]. From a functional point of view, S727 phosphorylation is mainly important for STAT3-dependent cellular survival and proliferation [43] modulating key targets such as Socs2, NanoG, and Klf4. In correlation to these assigned functions, it is not surprising that S727 phosphorylation is involved in v-Src-mediated transformation [38,39] and in c-Src/p42/p44 MAPK pathway-dependent breast cancer onset [26]. In addition, STAT3-dependent PCKε oncogenic action detected in some solid tumors is also mediated by S727 phosphorylation [44]. In conclusion, this PTM also shows promising characteristics for cancer therapy.
The STAT3 protein can also be phosphorylated in other residues of transcriptional activation domain (TAD), in C-terminal of sequence: S691 [53], T714 [54,55], T717 [55], and S719 [56]. Although mass spectrometry analysis has revealed phosphate groups in the residues just mentioned, no clear results about their functions in the regulation of STAT3 activities have been provided yet. For this reason, in this review, we decided to focus our attention only on Y705 and S727.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9080956
This entry is offline, you can click here to edit this entry!