In solid tumors, vasculogenic mimicry (VM) is the formation of vascular structures by cancer cells, allowing to generate a channel-network able to transport blood and tumor cells. While angiogenesis is undertaken by endothelial cells, VM is assumed by cancer cells. Besides the participation of VM in tumor neovascularization, the clinical relevance of this process resides in its ability to favor metastasis and to drive resistance to antiangiogenic therapy. VM occurs in many tumor types, including breast cancer, where it has been associated with a more malignant phenotype, such as triple-negative and HER2-positive tumors. The latter may be explained by known drivers of VM, like hypoxia, TGFB, TWIST1, EPHA2, VEGF, matrix metalloproteinases, and other tumor microenvironment-derived factors, which altogether induce the transformation of tumor cells to a mesenchymal phenotype with a high expression rate of stemness markers.
- vasculogenic mimicry
- breast cancer
- tumor neovascularization
1. What Is Vascular Mimicry?
Tumor growth and dissemination depend on vascularization, a process that is achieved through vasculogenesis and/or angiogenesis. Vasculogenesis is de novo blood vessel formation by newly differentiated endothelial cells (ECs), while angiogenesis is the formation of blood vessels from pre-existing ones, either by sprouting or intussusception [1]. Intussusception refers to the formation of pillars inside the blood vessel, resulting in its division into segments [2]. In the context of cancer, both vasculogenesis and angiogenesis are regulated by microenvironment-derived factors, tumor heterogeneity, cell–cell interactions (including malignant and non-transformed cells), as well as modifications on extracellular matrix (ECM) components [3]. In addition, there are alternative non-angiogenic mechanisms used by tumor cells to obtain nutrients and oxygen and to disseminate to distant sites, for instance vessel co-option, which consists in the hijacking of pre-existing blood vessels from non-tumoral surrounding tissue [4]. While all these processes and their related pathways play an essential role in the growth, proliferation, migration, invasion, and metastasis of highly aggressive tumors, they are not the only mechanisms by which tumors generate vasculature and escape routes [5][6]. In 1999, Maniotis and collaborators described for the first time an endothelial-independent vascularization mechanism in highly aggressive and metastatic uveal and cutaneous melanoma tumors. They observed the presence of patterned networks of interconnected loops and cord formation, composed of cancer cells and ECM that stained with the periodic acid-Schiff (PAS) reagent, but that in relation to EC markers such as CD31 or factor VIII-related antigen, showed weak, focal, and discontinuous staining. Erythrocytes could be found inside these structures, suggesting that they actually conducted blood and represented an intratumor microcirculatory system. Moreover, it was shown that highly invasive M619 human melanoma cells were able to form three-dimensional channel-like structures resembling vascular networks. This process was termed “vasculogenic mimicry” (VM) [7][8][9][10].
Vascular channels in VM share several characteristics with endothelial-dependent vasculature; however, distinctive features differentiate them (Table 1). For instance, ECs express vascular endothelial (VE)-Cadherin, also known as CD144, the major molecule related to cell–cell adhesion in endothelial adherent junctions. However, in cancer cells capable of forming VM, VE-Cadherin is aberrantly expressed and seems to be involved in a different function, namely, the acquisition of tubule-like structures [11]. Even if at present time there is no infallible biomarker for VM channels identification, some specific characteristics and the expression of particular markers associated with these cellular arrangements have been described (Table 1).
Table 1. Distinctive features/markers between vasculogenic mimicry and angiogenesis.
| Vasculogenic Mimicry | Angiogenesis | References |
|---|---|---|
| Formation of vascular channels from cancer stem cells (tumor cells). | Development of new blood vessels and capillaries from pre-existing ones. | [1][12] |
| Patterned networks of interconnected loops and cords formation | Formation by sprouting or intussusception | [1][7] |
| Formed by tumor cells and cancer stem cells | Formed by endothelial cells | [7] |
| Aberrant expression of VE-Cadherin | VE-Cadherin localization in cell membranes | [13] |
| PAS+, CD31−/low staining | PAS−/low, CD31+ staining | [7] |
| Factor VIII-related antigen negative or low | Factor VIII-related antigen highly positive | [7] |
| Unaffected by endostatin and other antiangiogenic factors | Inhibited by antiangiogenic factors | [14][15] |
| EPHA2, TIE1, LAMC2, overexpression | EPHA2, TIE1, LAMC2 generally negative. | [16] |
| Express stemness markers, e.g., CD133, ALDH1 | CD133 positivity mostly in endothelial precursor cells | [17][18][19][20] |
| More abundant in poorly differentiated tumors, such as HER2+ and TNBC | Present in embryogenesis, wound healing and tumor growth | [16][21] |
There are two types of VM described up to now. The tubular type and the patterned matrix type [22] (Figure 1). In vitro, the first type refers to networks of cellular cords above a thin matrix, encircling cell-free spaces (Figure 1a). In vivo, this type would appear as matrix “rivers” that may arrange as parallel PAS+ ECM deposits (Figure 1b). This matrix is produced by cancer cells. In some cases, PAS+ tumor endothelial-like cells can be found forming cords or lining blood channels. In the second type, PAS+ ECM patterns enclose packs of tumor cells wedged into the matrix arrays (Figure 1c,d). This last one is characteristic of highly invasive tumors [22]. It may be possible that the patterned type gives rise to the tubular type, after the enclosed cells die. In Figure 2, we provide photographs depicting VM-structures formed in vitro and in vivo by the triple-negative breast cancer (TNBC) cell line MBCDF-Tum, reported as highly tumorigenic [23] (Figure 2).
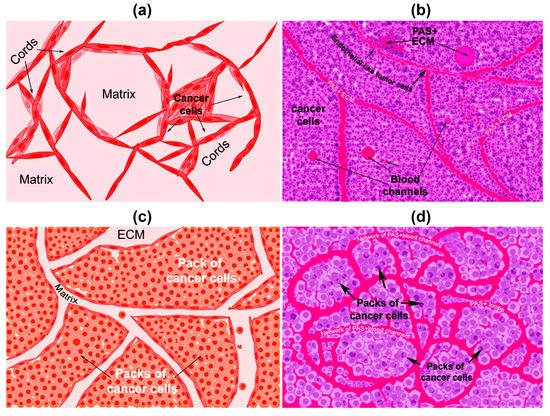
Figure 1. Graphical representation of the types of vasculogenic mimicry structures formed in vitro and in vivo by cancer cells. (a) In vitro tubular type. Is formed by networks of cellular cords encircling cell-free spaces above a matrix rich in collagen such as Matrigel. Tumor cells aligned in cords or tubular-like interconnected structures are depicted. (b) In vivo tubular type (parallel PAS+ patterned). Deposits of PAS+ proteoglycan/laminin-enriched matrix derived from cancer cells resembling “matrix rivers” may contain PAS+ tumor cells able to form channels and may be flanked by endothelial-like tumor cells. (c) In vitro patterned matrix type. Flattened tumor cells lodged into the matrix form packages of cells that deposit matrix enriched in collagen, laminin, and proteoglycans. Tumor endothelial-like cells may be found surrounding the packs. (d) In vivo patterned matrix type (network or back-to-back loops PAS+ patterned). Several layers of extracellular matrix rich in laminin, fibronectin, and collagens IV and VI form loops surrounding packs of tumor cells.
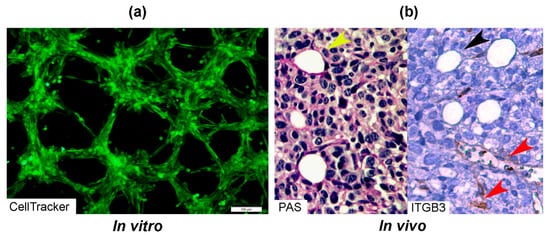
Figure 2. Photographs of vascular mimicry structures formed by the TNBC cell line MBCDF-Tum in vitro and in vivo. (a) MBCDF-Tum cells were labeled using Cell tracker-green (Abcam) and photographed by epifluorescence microscopy at 24 h of seeding (10 magnification, Olympus BX51). (b) MBCDF-Tum cells xenografted in nude mice generated VM-forming tumors. VM structures (yellow arrow heads) were identified by PAS-staining (magenta color, left side of the picture) in a tumor section. In the right part of the picture, a similar section of the same tumor was stained for Integrin-β3 (ITGB3) as an endothelial marker, identifying tumor endothelial-vasculature (brown staining, red arrow heads) in a hot spot of ITGB3-negative VM channels (black arrow heads) (40 magnification).
2. First Highlights of VM in Breast Cancer
VM has been identified in numerous types of highly aggressive tumors including breast cancer. Only two years later from the first report of VM in melanoma, a group in Japan identified the presence of blood pooling without a lining of ECs on hyper vascularized xenografts of inflammatory breast cancer. Remarkably, these cells were able to form tube-like structures and loops in vitro, and were associated with lung metastasis in vivo, representing the first evidence of VM in breast cancer [24]. These results helped to establish the relationship between angiogenesis and VM. Shirakawa et al. observed that the hyper vascularized zone in the tumor periphery contained vessels lined by ECs positive to murine CD31, consistent with angiogenesis, while the central highly hypoxic area of the tumor exhibited channels that were PAS positive, presented weak expression of human integrin αvβ3 and lacked ECs, consistent with VM. Altogether, this suggested that in some instances, tumors can develop hybrid vascular networks combining angiogenesis and VM to efficiently obtain oxygen and nutrients [25][26]. In addition, structural heterogeneity (mosaic vessels) has also been described in solid tumors, including breast cancer, where a vessel may be lined by ECs in some parts and by tumor cells in others, forming hybrid vascular structures associated with intravasation and systemic dissemination of cancer cells [27]. Since it has been demonstrated that VM can enhance metastasis after an anti-angiogenic treatment [28], research in the VM field will surely improve cancer therapeutics.
3. Clinical Relevance of VM in Breast Cancer and Association with Clinicopathological Parameters
There is no doubt that a major drawback of anti-angiogenic treatment is the formation of VM. Indeed, by inducing hypoxia, VM may be favored, which in turn enhances distant metastasis [28][29]. Notably, the angiogenesis inhibitor endostatin readily inhibits proangiogenic factors such as vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF2), matrix metalloproteinases (MMPs), and hypoxia-inducible factor 1-α (HIF1A), blocking endothelial tube formation. However, endostatin does not affect VM-forming cells, which after being exposed to this collagen-derived factor remain fully active and capable to configure vascular channels [14]. Other antiangiogenic factors have shown similar results [15], suggesting a differential response of EC-dependent angiogenesis and cancer-dependent VM channels formation. In addition, as with the well-established relationship between microvascular density and metastasis in invasive breast cancer [30], VM also has been associated with malignant cells dissemination and bad prognosis, including higher recurrence, lower survival, larger tumor size, and poorer differentiation grade [16][25][31][32], linking this feature to a more malignant breast cancer phenotype [25][31][32]. The VM-positivity rate and its impact on clinicopathological parameters and prognosis in breast cancer patients have been largely studied in the last two decades. For instance, the study from Shirakawa K et al. [25] showed that from 331 surgically resected breast cancer specimens, only 26 (7.9%) evidenced the presence of VM. A high proportion of these VM-positive tumors exhibited pseudo-comedo formations, which are channels containing blood cells instead of necrotic tumor cells. Notably, in these 26 cases, patients were more likely to have hematogenous recurrence and lower percentage of 5-year survival [25]. However, in another study involving eight clinical reports with 1238 breast cancer patients, the VM-cases rate was higher, specifically 24%, and this was associated with larger tumor size (>2 cm), lymph node metastasis, poorer differentiation grade (grades 2 and 3), and shorter overall survival than those without VM, corroborating that this feature is associated with more aggressive breast cancer tumors and poorer prognosis [32].
Interestingly, in invasive ductal carcinoma samples, VM was detected in 13.3% of the analyzed tumors. Still, in this VM-positive group, 75% were significantly associated with bad clinicopathological characteristics, including axillary lymph node metastasis (95.6%), tumor size larger than 3 cm (56.25%), higher histological grade (stage 3, 75%), and overall poor prognosis [33]. Similarly, another study showed that breast cancer patients with VM-positive tumors were related to positive nodal status and advanced clinical stage, being the majority of VM-cases in progressive stage 2 and 3, thus, again, associating VM to a poorer outcome [34]. Of note, a meta-analysis on the role of VM in cancer progression and its prognostic value was undertaken considering different types of tumors, corroborating that the presence of VM predicts poorer survival outcomes in cancer patients [35].
4. Relationship between VM and Tumor Phenotype
Human breast cancer tumors are classified mainly considering clinic and histopathologic features, as well as molecular markers. Regarding this, the vast majority of these tumors belong to a group that expresses estrogen receptor alpha (ERα) and progesterone receptor (PR). Tumors overexpressing epidermal growth factor receptor 2 (HER2) generally lack ER and PR, while those that do not express neither of these three proteins are collectively called TNBC tumors. HER2 and TNBC are commonly considered as the most aggressive phenotypes of breast cancer.
The association between VM and breast tumor phenotype has been investigated. In vitro studies have shown that TNBC aggressive cells are particularly prone to form tubular structures, in contrast to more differentiated breast cancer cells. For example, the TNBC MDA-MB-231 and HCC1937 cells readily formed tubular-like structures in Matrigel [36][37]. In comparison, the ERα-positive cell line MCF-7 has been reported to be incapable of forming VM in this matrix [36]; however, in the presence of some VM drivers such as interleukin 1β, MCF-7 cells formed microvessel-like intersections and cords [38]. Further studies are needed to corroborate the effects of VM-drivers upon tubular-like structure formation in ER-positive breast cancer cells.
The link between VM and a more malignant breast cancer phenotype is coherent with the previously discussed association between VM and poor prognosis, as well as with the stemness features and increased plasticity characterizing cells with high VM-forming potential [18][39]. Indeed, some stemness markers have been negatively related to the hormone receptor status, while their expression has been found significantly increased in TNBC [18][39]. There are important features of the genotypic and phenotypic differences in breast cancer that confer a greater capacity to develop VM, like in TNBC compared to hormone receptor-positive or HER2-positive tumors. For instance, BRCA1 mutations have been shown to predispose for the basal-like/TNBC tumor subtype [40].
On the other hand, there is also solid evidence showing a positive association between VM and the overexpression of HER2. In a study using the MCF-7 cells, forced exogenous HER2 overexpression allowed these cells to form vessel-like structures in Matrigel, a characteristic previously absent in the parental cell line. Interestingly, this process was associated with increased VE-Cadherin protein expression, which abundance and interaction with the epithelial cell kinase 2 (EPHA2) are known to be linked to VM induction [34][41]. Strongly supporting these observations, studies undertaken in aggressive melanoma cells have shown that knockdown or downregulation of VE-cadherin, EPHA2, or laminin subunit gamma 2 (LAMC2) results in abolishing of their ability to form VM [42][43][44]. Notably, in invasive breast carcinoma specimens, HER2 overexpression highly correlated with VM, further corroborating the in vitro results in MCF-7 cells [33][34]. However, other studies have not found a statistically significant association between HER2 overexpression and VM [32]. The reason for this discrepancy is not known, but may be related to an incomplete transformation to a full vasculogenic phenotype in HER2-positive cells, probably due to a lesser level of aggressiveness or the development of alternative survival pathways not related to HER2. Supporting this hypothesis, it is known that HER2-positive tumor cells previously treated with trastuzumab express antigens normally associated with endothelial and stemness phenotypes, together with VM markers, indicating that the treatment may induce VM. However, and interestingly, these cells were not able to form VM structures unless they had fully developed resistance to trastuzumab. Indeed, trastuzumab-resistant cells readily formed tubular structures on Matrigel, which suggested that while HER2-positive cells remain sensitive to treatment, an incomplete vasculogenic phenotype prevails, while fully resistant cells have already experienced a complete transformation and therefore can form VM channels [45].
This entry is adapted from the peer-reviewed paper 10.3390/cells10071758
References
- Kolte, D.; McClung, J.A.; Aronow, W.S. Chapter 6—Vasculogenesis and Angiogenesis. In Translational Research in Coronary Artery Disease; Aronow, W.S., McClung, J.A., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 49–65.
- Patan, S.; Munn, L.L.; Jain, R.K. Intussusceptive Microvascular Growth in a Human Colon Adenocarcinoma Xenograft: A Novel Mechanism of Tumor Angiogenesis. Microvasc. Res. 1996, 51, 260–272.
- Rodríguez-Núñez, I.; Romero, F.; González, M.; Campos, R.R. Biología del Desarrollo Vascular: Mecanismos en Condiciones Físiológicas y Estrés Flujo. Int. J. Morphol. 2015, 33, 1348–1354.
- Kuczynski, E.A.; Vermeulen, P.B.; Pezzella, F.; Kerbel, R.S.; Reynolds, A.R. Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 469–493.
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Reviews Cancer 2003, 3, 401–410.
- Folberg, R.; Hendrix, M.J.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381.
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752.
- Folberg, R.; Arbieva, Z.; Moses, J.; Hayee, A.; Sandal, T.; Kadkol, S.; Lin, A.Y.; Valyi-Nagy, K.; Setty, S.; Leach, L.; et al. Tumor cell plasticity in uveal melanoma: Microenvironment directed dampening of the invasive and metastatic genotype and phenotype accompanies the generation of vasculogenic mimicry patterns. Am. J. Pathol. 2006, 169, 1376–1389.
- Basu, G.D.; Liang, W.S.; Stephan, D.A.; Wegener, L.T.; Conley, C.R.; Pockaj, B.A.; Mukherjee, P. A novel role for cyclooxygenase-2 in regulating vascular channel formation by human breast cancer cells. Breast Cancer Res. 2006, 8, 69.
- Chiao, M.T.; Yang, Y.C.; Cheng, W.Y.; Shen, C.C.; Ko, J.L. CD133+ glioblastoma stem-like cells induce vascular mimicry in vivo. Curr. Neurovascular Res. 2011, 8, 210–219.
- Delgado-Bellido, D.; Serrano-Saenz, S.; Fernández-Cortés, M.; Oliver, F.J. Vasculogenic mimicry signaling revisited: Focus on non-vascular VE-cadherin. Mol. Cancer 2017, 16, 65.
- Andonegui-Elguera, M.A.; Alfaro-Mora, Y.; Caceres-Gutierrez, R.; Caro-Sanchez, C.H.S.; Herrera, L.A.; Diaz-Chavez, J. An Overview of Vasculogenic Mimicry in Breast Cancer. Front. Oncol. 2020, 10, 220.
- Breier, G.; Grosser, M.; Rezaei, M. Endothelial cadherins in cancer. Cell Tissue Res. 2014, 355, 523–527.
- Hendrix, M.J.; Seftor, E.A.; Hess, A.R.; Seftor, R.E. Vasculogenic mimicry and tumour-cell plasticity: Lessons from melanoma. Nat. Rev. Cancer 2003, 3, 411–421.
- van der Schaft, D.W.; Seftor, R.E.; Seftor, E.A.; Hess, A.R.; Gruman, L.M.; Kirschmann, D.A.; Yokoyama, Y.; Griffioen, A.W.; Hendrix, M.J. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J. Natl. Cancer Inst. 2004, 96, 1473–1477.
- Mitra, D.; Bhattacharyya, S.; Alam, N.; Sen, S.; Mitra, S.; Mandal, S.; Vignesh, S.; Majumder, B.; Murmu, N. Phosphorylation of EphA2 receptor and vasculogenic mimicry is an indicator of poor prognosis in invasive carcinoma of the breast. Breas Cancer Res Treat. 2020, 179, 359–370.
- Zhang, D.; Sun, B.; Zhao, X.; Ma, Y.; Ji, R.; Gu, Q.; Dong, X.; Li, J.; Liu, F.; Jia, X.; et al. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer. Mol. Cancer 2014, 13, 207.
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632.
- Liu, T.J.; Sun, B.C.; Zhao, X.L.; Zhao, X.M.; Sun, T.; Gu, Q.; Yao, Z.; Dong, X.Y.; Zhao, N.; Liu, N. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 2013, 32, 544–553.
- Rossi, E.; Poirault-Chassac, S.; Bieche, I.; Chocron, R.; Schnitzler, A.; Lokajczyk, A.; Bourdoncle, P.; Dizier, B.; Bacha, N.C.; Gendron, N.; et al. Human Endothelial Colony Forming Cells Express Intracellular CD133 that Modulates their Vasculogenic Properties. Stem Cell Rev Rep. 2019, 15, 590–600.
- Ribatti, D.; Pezzella, F. Overview on the Different Patterns of Tumor Vascularization. Cells 2021, 10, 639.
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. Acta Pathol. Microbiol. Et Immunol. Scand. 2004, 112, 508–525.
- Garcia-Quiroz, J.; Garcia-Becerra, R.; Santos-Cuevas, C.; Ramirez-Nava, G.J.; Morales-Guadarrama, G.; Cardenas-Ochoa, N.; Segovia-Mendoza, M.; Prado-Garcia, H.; Ordaz-Rosado, D.; Avila, E.; et al. Synergistic Antitumorigenic Activity of Calcitriol with Curcumin or Resveratrol is Mediated by Angiogenesis Inhibition in Triple Negative Breast Cancer Xenografts. Cancers 2019, 11, 1739.
- Shirakawa, K.; Tsuda, H.; Heike, Y.; Kato, K.; Asada, R.; Inomata, M.; Sasaki, H.; Kasumi, F.; Yoshimoto, M.; Iwanaga, T.; et al. Absence of endothelial cells, central necrosis, and fibrosis are associated with aggressive inflammatory breast cancer. Cancer Res. 2001, 61, 445–451.
- Shirakawa, K.; Wakasugi, H.; Heike, Y.; Watanabe, I.; Yamada, S.; Saito, K.; Konishi, F. Vasculogenic mimicry and pseudo-comedo formation in breast cancer. Int. J. Cancer 2002, 99, 821–828.
- Shirakawa, K.; Kobayashi, H.; Heike, Y.; Kawamoto, S.; Brechbiel, M.W.; Kasumi, F.; Iwanaga, T.; Konishi, F.; Terada, M.; Wakasugi, H. Hemodynamics in vasculogenic mimicry and angiogenesis of inflammatory breast cancer xenograft. Cancer Res. 2002, 62, 560–566.
- Silvestri, V.L.; Henriet, E.; Linville, R.M.; Wong, A.D.; Searson, P.C.; Ewald, A.J. A Tissue-Engineered 3D Microvessel Model Reveals the Dynamics of Mosaic Vessel Formation in Breast Cancer. Cancer Res. 2020, 80, 4288–4301.
- Xu, Y.; Li, Q.; Li, X.Y.; Yang, Q.Y.; Xu, W.W.; Liu, G.L. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J. Exp. Clin. Cancer Res. 2012, 31, 1–7.
- Valencia-Cervantes, J.; Huerta-Yepez, S.; Aquino-Jarquín, G.; Rodríguez-Enríquez, S.; Martínez-Fong, D.; Arias-Montaño, J.A.; Dávila-Borja, V.M. Hypoxia increases chemoresistance in human medulloblastoma DAOY cells via hypoxia-inducible factor 1α-mediated downregulation of the CYP2B6, CYP3A4 and CYP3A5 enzymes and inhibition of cell proliferation. Oncol. Rep. 2019, 41, 178–190.
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N. Engl. J. Med. 1991, 324, 1–8.
- Sun, B.; Zhang, S.; Zhang, D.; Du, J.; Guo, H.; Zhao, X.; Zhang, W.; Hao, X. Vasculogenic mimicry is associated with high tumor grade, invasion and metastasis, and short survival in patients with hepatocellular carcinoma. Oncol. Rep. 2006, 16, 693–698.
- Shen, Y.; Quan, J.; Wang, M.; Li, S.; Yang, J.; Lv, M.; Chen, Z.; Zhang, L.; Zhao, X.; Yang, J. Tumor vasculogenic mimicry formation as an unfavorable prognostic indicator in patients with breast cancer. Oncotarget 2017, 8, 56408–56416.
- Jafarian, A.H.; Kooshkiforooshani, M.; Rasoliostadi, A.; Mohamadian Roshan, N. Vascular Mimicry Expression in Invasive Ductal Carcinoma; A New Technique for Prospect of Aggressiveness. Iran. J. Pathol. 2019, 14, 232–235.
- Liu, T.; Sun, B.; Zhao, X.; Gu, Q.; Dong, X.; Yao, Z.; Zhao, N.; Chi, J.; Liu, N.; Sun, R.; et al. HER2/neu expression correlates with vasculogenic mimicry in invasive breast carcinoma. J. Cell. Mol. Med. 2013, 17, 116–122.
- Yang, J.P.; Liao, Y.D.; Mai, D.M.; Xie, P.; Qiang, Y.Y.; Zheng, L.S.; Wang, M.Y.; Mei, Y.; Meng, D.F.; Xu, L.; et al. Tumor vasculogenic mimicry predicts poor prognosis in cancer patients: A meta-analysis. Angiogenesis 2016, 19, 191–200.
- Quiros-Gonzalez, I.; Tomaszewski, M.R.; Aitken, S.J.; Ansel-Bollepalli, L.; McDuffus, L.A.; Gill, M.; Hacker, L.; Brunker, J.; Bohndiek, S.E. Optoacoustics delineates murine breast cancer models displaying angiogenesis and vascular mimicry. Br. J. Cancer 2018, 118, 1098–1106.
- Luan, Y.Y.; Liu, Z.M.; Zhong, J.Y.; Yao, R.Y.; Yu, H.S. Effect of grape seed proanthocyanidins on tumor vasculogenic mimicry in human triple-negative breast cancer cells. Asian Pac. J. Cancer Prev. 2015, 16, 531–535.
- Nisar, M.A.; Zheng, Q.; Saleem, M.Z.; Ahmmed, B.; Ramzan, M.N.; Ud Din, S.R.; Tahir, N.; Liu, S.; Yan, Q. IL-1β Promotes Vasculogenic Mimicry of Breast Cancer Cells Through p38/MAPK and PI3K/Akt Signaling Pathways. Front. Oncol. 2021, 11, 1–13.
- Xing, P.; Dong, H.; Liu, Q.; Zhao, T.; Yao, F.; Xu, Y.; Chen, B.; Zheng, X.; Wu, Y.; Jin, F.; et al. ALDH1 Expression and Vasculogenic Mimicry Are Positively Associated with Poor Prognosis in Patients with Breast Cancer. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 961–970.
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423.
- Hess, A.R.; Seftor, E.A.; Gruman, L.M.; Kinch, M.S.; Seftor, R.E.; Hendrix, M.J. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: Implications for vasculogenic mimicry. Cancer Biol. Ther. 2006, 5, 228–233.
- Hess, A.R.; Seftor, E.A.; Gardner, L.M.; Carles-Kinch, K.; Schneider, G.B.; Seftor, R.E.; Kinch, M.S.; Hendrix, M.J. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: Role of epithelial cell kinase (Eck/EphA2). Cancer Res. 2001, 61, 3250–3255.
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023.
- Seftor, R.E.; Seftor, E.A.; Koshikawa, N.; Meltzer, P.S.; Gardner, L.M.; Bilban, M.; Stetler-Stevenson, W.G.; Quaranta, V.; Hendrix, M.J. Cooperative interactions of laminin 5 gamma2 chain, matrix metalloproteinase-2, and membrane type-1-matrix/metalloproteinase are required for mimicry of embryonic vasculogenesis by aggressive melanoma. Cancer Res. 2001, 61, 6322–6327.
- Hori, A.; Shimoda, M.; Naoi, Y.; Kagara, N.; Tanei, T.; Miyake, T.; Shimazu, K.; Kim, S.J.; Noguchi, S. Vasculogenic mimicry is associated with trastuzumab resistance of HER2-positive breast cancer. Breast Cancer Res. 2019, 21, 88.