1. Inflammation and ECM Remodeling, Close Allies in Cancer Progression
In 1986, Dvorak defined cancers as “wounds that do not heal”. This definition comes from the observation that tumor growth leads to a continuous injury of the surrounding stromal tissue that triggers, in turn, a chronic response in the attempt to repair the lesion and restore tissue homeostasis [
10]. This response is very complex and multifaceted and includes the concerted activity of inflammatory molecules, immune cells, and fibroblasts, which together lead to a state of persistent inflammation.
When a tumor lesion occurs, immune cells, as leukocytes, macrophages, and/or bone marrow-derived myeloid precursors, are recruited at the injured site. These tumor-infiltrating cells, along with cancer cells, release in the TME high levels of transforming growth factor β (TGFβ), a cytokine that exerts pleiotropic effects on both cancer and normal cells adjacent to rupture through autocrine and paracrine signaling mechanisms [
11].
In normal and premalignant stages of cell transformation, TGFβ exerts tumor-inhibiting functions by suppressing cancer cell growth through anti-proliferative and pro-apoptotic signaling [
12]. Moreover, TGFβ limits the proliferation and differentiation of cells of the innate and adaptive immune system, thus suppressing tumorigenic inflammation. However, at more advanced stages of carcinogenesis, tumor cells acquire inactivating mutations of TGFβ receptors and/or alteration of their signaling transduction pathway (reviewed in [
12]) and become resistant to the negative regulatory effects of this cytokine. As a consequence, TGFβ switches from a tumor suppressor factor to a metastasis promoter and drives changes in the TME that finally sustain tumor growth [
13]. Indeed, high levels of TGFβ in the TME enable cancer cells to escape the immune surveillance leading to an over-production of cytokines and chemokines that contribute to boosting chronic inflammation [
12,
13]. Moreover, TGFβ produced by cancer cells acts in a paracrine way on stromal cells stimulating the secretion of growth factors and mitogens, as the platelet-derived growth factor (PDGF), and driving the trans-differentiation of stromal progenitors, including resident fibroblasts, endothelial cells, preadipocytes, and bone marrow-derived mesenchymal stem cells (MSCs) into “activated fibroblast” [
14,
15,
16]. These “activated fibroblasts”, collectively named CAFs, bear a myofibroblast-like phenotype, express higher levels of αSMA, collagen 11-α1 (COL11A1), PDGF receptor (PDGFR) α/β, and lower levels of caveolin-1 than normal fibroblasts, and acquire greater contractile and proliferating capacities [
17].
However, to date, specific markers to outline the molecular profile of CAFs are still lacking, making it difficult to better understand the biology of these heterogeneous classes of cells [
18]. Once activated, CAFs themselves release TGFβ and feed an autocrine-positive signaling loop that sustains tumor progression by contributing to the generation of a permissive TME [
19], as illustrated in
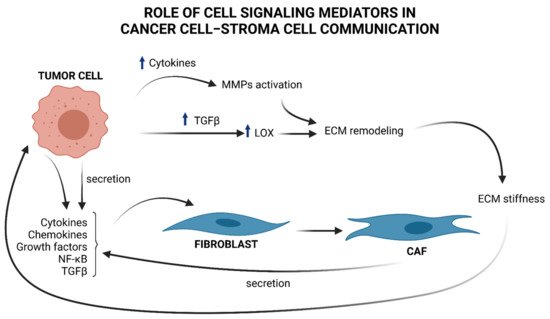
Figure 1.
Figure 1. Brief overview of cell signaling mediators in cancer cell–stroma cell communication and ECM remodeling. Tumor cells produce cytokines, chemokines, NF-κB, and TGFβ, that “activate” stromal progenitors, such as fibroblasts, into CAFs and produce, in turn, cytokines and TGFβ, influencing tumor cell functions. TGFβ upregulation in cancer cells provokes an increment in LOX, which in turn remodels ECM by favoring collagen fibers cross-linking. As a consequence ECM stiffness increases and induces the mechanical activation of latent TGFβ. Cytokines produced by tumor cells contributes to MMPs activation. These enzymes take part to ECM remodeling and sustains tumor progress.
Under TGFβ stimulation, CAFs release high levels of cytokines and chemokines, contributing to attracting neutrophils, macrophages, lymphocytes, and natural killer (NK) cells at the site of the tumor lesion, thus boosting the inflammatory response that favors cancer progression [
16]. One of the best characterized pro-tumorigenic cytokines is interleukin 6 (IL-6), known to be linked to increased risk of development of a large variety of cancers [
20]. IL-6, secreted by CAFs, binds to its cognate receptor expressed by tumor cells and promotes their growth and the acquisition of an invasive phenotype by activating the IL-6–JAK–STAT axis and the notch pathway [
21]. IL-6 secreted from cancer cells also acts in a paracrine manner to induce the differentiation of fibroblasts into CAFs. By these efferent (from tumor to stroma) and afferent (from stroma to tumor) signaling pathways, IL-6 is one of the molecules secreted in the TME that participates in the tumor–stroma cross-talk required for tumor growth [
22].
Tumor necrosis factor-α (TNFα) is another cytokine secreted at high levels in the TME during the early stages of tumorigenesis that has an established role in chronic inflammation, angiogenesis, tissue remodeling, tumor growth, and metastasis [
23]. Similarly to TGFβ, the role played by TNFα in cancer progression is both tumor-inhibiting and tumor-promoting, depending on the cell context and the cancer stage. Indeed, although high levels of TNFα inhibit tumor growth [
24], secretion of low levels of TNFα by cancer cells in a B16 mouse melanoma model furthers the recruitment of infiltrating myeloid cells, promoting cancer vascularization and progression [
25]. Furthermore, in a mouse xenograft model of ovarian carcinoma, TNFα receptor 1 (TNFR1) expression on CD4+ T cells is necessary for IL-17 secretion and myeloid cell recruitment in tumors, sustaining inflammation and cancer progression [
26].
Apart from participating in the complex signaling between tumor and surrounding stroma, CAFs are also responsible for the deposition of ECM components (see
Figure 1), a physiological process that becomes highly unregulated in the early stages of cancer [
27]. This dysregulation compromises abundance, concentration, structure, and architecture of the ECM, affecting tumor establishment and progression. For example, deposition of collagen by CAFs, together with changes in the expression of the enzymes responsible for its remodeling, leads to an impairment of the organization of collagen fibers and results in increased stiffness of the ECM [
28]. In breast cancer, the collagen fibers become linearized and perpendicularly oriented at the tumor boundary, thus favoring the migration and invasion of cancer cells [
29]. The lysyl oxidases (LOX) enzymes are responsible for the cross-linking of collagen and elastin fibers. The expression of LOX is upregulated in many cancers by a mechanism that is, at least in part, TGFβ-driven [
30]. An unbalanced enhancement in LOX expression and activity increases ECM stiffness that induces the mechanical activation of latent TGFβ and fuels the onset of a vicious circle that maintains a tumor-promoting inflammatory environment [
28]. The activity of LOX enzymes is therefore not restricted to the shaping of the architecture and the mechanical functions of the ECM but is part of a complex network of bi-directional signaling within the matrix and different cells of the microenvironment that contributes to establish and maintain an inflamed, immune-suppressive, and pro-carcinogenic TME [
30].
The activity of ECM-degrading proteases, for example, releases matrix-bound growth factors and cytokines, which can mediate the activation of downstream effectors, such as various oncogenic transcription factors [
11,
27].
Among these, NF-κB is a prototypical inflammatory mediator involved in tumorigenesis. NF-κB transient activation is tightly controlled under physiological conditions that promote inflammation, as an adaptive, physiological response. However, in cancer proinflammatory cytokines, oncogenic growth factors and tyrosine kinases promote the constitutive activation of NF-κB through autocrine and paracrine ways [
31]. This persistent activation yields to the transcriptional regulation of various ECM components and ECM degrading enzymes, such as MMPs, whose expression is under the control of NF-κB [
32].
To degrade the ECM, different types of cells which constitute the tumor mass, i.e., the actual cancer cells, activated fibroblasts, and macrophages, produce a large number of enzymes, belonging to the metzincin protease superfamily of zinc-endopeptidases, which include MMPs, also known as matrixins, a disintegrin and metalloproteinases (ADAMs), and ADAMs with thrombospondin motifs (ADAMTSs).
MMPs induced by pro- and inflammatory factors and regulated by NF-κB [
32], as mentioned above, have an important role in sustaining all six major hallmarks of cancer [
33] highlighted within the complexity of cancer biology: (1) sustaining proliferative signaling; (2) evasion of apoptosis; (3) angiogenesis and lymphangiogenesis; (4) invasion and metastasis; (5) reprogramming of energy metabolism; and (6) evasion of the immune response. Indeed, there is a close relationship between the release of cytokines and the specific ECM-degrading enzymes MMPs, as MMPs may play a non-proteolytic and non-ECM role in cell–cell communication [
34] (
Figure 1). For a deeper description of the role of specific MMPs regulated by cytokines, see Review [
34].
2. Extracellular Chaperones in the TME: Focus on Their Roles in Cancer-Related Inflammation and ECM Remodeling
The protein homeostasis of cancer cells is challenged by an increased rate of protein synthesis that is required to face the anabolic demand of fast-growing and proliferating cells. Oxygen and nutrients limitations drive changes in the metabolism of cancer cells that contribute to the maintenance of an acidified, oxidized, and inflamed TME favoring the unfolding of many extracellular proteins [
97].
As a consequence, cancer cells upregulate the activity of their protein quality systems that collectively constitute the proteostasis network in the attempt to reduce the proteotoxic stress and keep the proteome as stable and functional as possible [
98].
Molecular chaperones are first-line players of the proteostasis network due to their ubiquitous expression and their ability to stabilize proteins in their native conformation, re-fold denatured polypeptides, inhibit unfolded protein aggregation, and direct misfolded proteins to proteasomal and autophagic degradation [
99,
100]. The wide and heterogeneous family of molecular chaperones comprises, among others, the heat shock proteins (HSPs), a family of stress-proteins that are induced in response to physical, chemical, and biological insults to support cell survival [
101]. According to their molecular weight, HSPs are currently classified into six groups. HSP100s, HSP90s, HSP70s, and HSP60s use the energy derived from ATP hydrolysis to assist protein folding. HSP40s and other small HSPs do not possess ATPase activity and act as a co-chaperone to HSP70s, binding to unfolded proteins preventing their aggregation and eventually delivering those irreparably damaged to the protein degradation systems [
102].
It is now widely accepted that many HSPs, initially classified as intracellular proteins, can reach the extracellular space through various mechanisms, including exocytosis of lysosomal vesicles, translocation through the plasma membrane, and release of extracellular vesicles as exosomes [
103,
104,
105,
106].
The function of extracellular HSPs (eHSP) in the TME is not limited to the canonical function of stress proteins that counteract protein unfolding (reviewed in Richter K. et al.) [
107]. More recent studies highlighted that eHSPs take part in the complex signaling network that influences the fate of cancer cells with different outcomes depending on the stage of the pathology [
101]. They counteract tumor growth due to their ability to bind antigenic peptides and activate anti-tumor innate immunity [
108], thus promoting immune surveillance and inhibiting inflammation [
109]. However, they favor tumor progression and metastasis formation by sustaining cancer proliferation, cell migration and invasion, neo-angiogenesis, and immune escape [
101].
The eHSPs interact directly with cell surface receptors and consequently engage intracellular signaling pathways that influence cell behavior [
101]. Low-density lipoprotein receptor-related protein 1 (LRP1) is a common receptor for many extracellular chaperones, both in normal and cancer cells where its expression is often upregulated [
110]. eHSP90 following LRP1 binding activates both AKT and NF-κB signaling, promoting cell proliferation, EMT, and ECM remodeling [
111,
112]. Similarly, various eHSPs activate Toll-like receptors (TLRs), induce pro-inflammatory signaling cascades through NF-κB and ERK activation, and promote tumor establishment releasing pro-inflammatory, anti-apoptotic, proliferative, and pro-fibrogenic signals on cancer cells and TME cells [
113].
Many ECM-degrading enzymes depend on eHSPs binding for their activity and stability. For instance, in breast cancer, eHSP90α mediates the proteolytic activation of MMP2 in cooperation with eHSP70 and other co-chaperones [
114]. eHSP90α interacts with lysyl oxidase-like 2 (LOXL2) and allows the latter to assume its functional conformation favoring the migration of breast cancer cells [
115] and with tissue plasminogen activator (tPA), favoring the conversion of plasminogen in plasmin, a process that favors cancer invasion [
116]. In breast cancer cells, both HSP90α and HSP90β are closely associated in a complex with fibronectin, participating in its assembly and turnover [
117,
118].