These ketimines are less electrophilic and thus, arguably, more challenging substrates. 3-Aryl-3-hydroxyisoindolin-1-ones are often employed as stable precursors for the corresponding endocyclic N-carbonyl diaryl ketimines. This class of ketimines is useful synthons to access chiral isoindolin-1-ones that are an important motif found in numerous biologically relevant molecules and natural products.
- ketimine Mannich
- ketimine allylation
- aza-Morita–Baylis–Hillman
- asymmetric catalysis
- α-tertiary amine
- β-amino carbonyl
1. Introduction
The condensation reaction between in situ generated imines and enols to form β-amino carbonyl compounds was reported by Carl U. F. Mannich for the first time in 1912 [1]. Since it provides direct access to synthetically useful chiral building blocks from readily available carbonyls and amines, it attracted huge attention from the synthetic community and is now recognized as one of the most important chemical transformations (selected reviews; In order to circumvent the inherent difficulties associated with the classical Mannich reaction such as regio-, stereo-, and product-selectivities, preformed imines and/or enolates have often been employed.
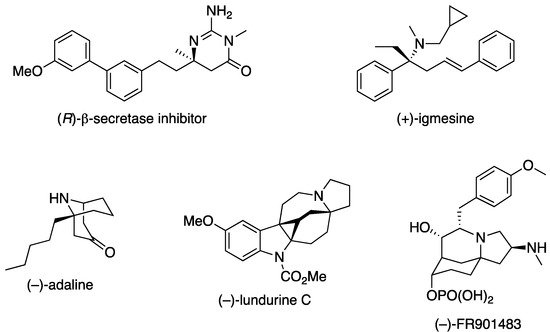
Over the years, the asymmetric catalytic Mannich reaction of ketimines (Scheme 1) has been of significant interest to synthetic and medicinal chemists as a way to access synthetically versatile compounds bearing an α-tertiary amine stereogenic center (selected reviews; [2][3][4][5][6][7][8][9][10][11][12]). It is because optically pure chiral α-tertiary amines are a key structural motif found in a large number of biologically relevant molecules and natural products (Figure 1) However, ketimines are much more challenging electrophiles than aldimines for the Mannich and its related reactions (e.g., allylation) due to some notorious structural properties of the formers (Figure 2). In general, diaryl, aryl alkyl, and dialkyl ketimines (i.e., unmodified ketimines) are poor electrophiles because of the severe steric demands and the electron-donating nature of the two groups flanking a C=N bond.
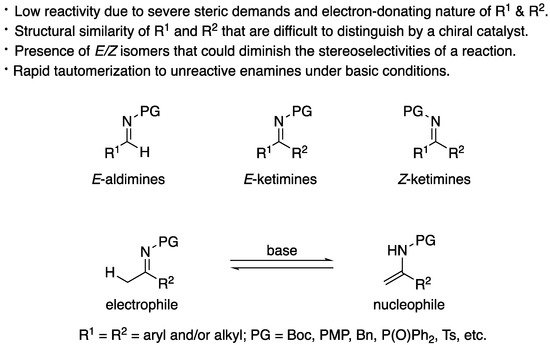
Nonendocyclic ketimines can equilibrate between theEandZforms in solution at room temperature by an inversion, or rotation, and furthermore, through ketimine–enamine tautomerization, if they have an α-hydrogen atom [13]. Therefore, ketimines are often available only as a mixture ofEandZisomers that could lead to diminished stereoselectivities in the asymmetric transformations. regardless of theE/Zisomeric ratio of the ketimine used. Despite this observation, it remains largely elusive how to logically develop such catalytic enantioselective methods.
The majority of the work published on the ketimine Mannich reaction and its related transformations employed ketimines where some notorious properties of unmodified ketimines summarized in Figure 2 were absent. In this mini-review, unmodified ketimines refer to nonendocyclic ketimines in which no electron-withdrawing groups and/or alkynes are attached to the carbon atom of a C=N bond (sp hybridized carbon atoms are electronegative; for example, the pKavalue of acetylene is 24 in H2O). It is arguable whether ketimines bearing strongly electron-withdrawingN-protecting groups such asN-tosyl ketimines should be categorized as unmodified ketimines or not; however, they are also included as part of unmodified ketimines as long as those ketimines are tautomerizable. We hope the information provided herein promotes the study and use of ketimines that are in general considered problematic substrates.
With respect to the transformations related to the ketimine Mannich reaction, only methods that employ enolates or its equivalents (e.g., silyl enol ethers and allyl metals) as nucleophiles to provide β-amino carbonyl compounds or its equivalents are covered, while vinylogous Mannich reactions are included. The asymmetric catalytic methods of ketimines that provide α-amino carbonyl compounds such as Strecker reaction (selected review; [14]), nitro-Mannich reaction (selected review; [15]), aza-benzoin reaction (selected references [16][17]) are not included. The asymmetric umpolung addition of ketimines to electrophiles (selected references; [18][19][20][21][22][23][24][25]) is beyond the scope of this review. Mannich reaction is an approach to avoid some drawbacks of preformed ketimines by in situ oxidation of corresponding amines, which is recently reviewed [26].
2. Unmodified Ketimines in the Mannich Reaction
Unless the ketimine activating groups are part of the desired products (e.g., CF3, isatin), they need to be transformed adequately or removed from the Mannich products, which adds steps that would not have been necessary otherwise (vide supra). Therefore, arguably, the development of the Mannich reaction with unmodified ketimines would be highly beneficial from the viewpoints of both atom and step economies [27][28][29].
In 2007, Kanai, Shibasaki, and coworkers disclosed the first catalytic enantioselective Mannich reaction of unmodified ketimines using a preformed silyl ketene acetal and chiral copper complexes [30]. In these reactions,N-di(3,5-xylyl)phosphinoyl ketimines were employed, and a highly nucleophilic copper enolate was generated from a silyl ketene acetal and a chiral copper complex through transmetalation. While optimal chiral ligands for aromatic and aliphatic ketimines turned out to be different, the method tolerated both classes of ketimines very well, giving the β,β-disubstituted amino acid equivalents in 61–92% yields with 91–97% ee and 45–99% yields with 58–81% ee, respectively. While they mentioned that the nucleophile scope was limited to acetate donors since α-substituted enolates did not undergo the addition reaction, this work still remains as the state-of-the-art catalytic enantioselective Mannich reaction between unmodified ketimines and acetate donors.
In 2013, Nakamura and coworkers reported a diastereo- and enantio-selective vinylogous Mannich reaction ofN-diphenylphosphinoyl ketimines and 2-(trimethylsiloxy)furan catalyzed by a chiral copper(II) complex (Scheme 28) [31]. They proposed that a nucleophilic chiral copper dienolate complex generated from 2-(trimethylsiloxy)furan was an actual nucleophilic species. Cinchona alkaloid-derived amide31was found to be an optimal ligand, and its copper (II) complex tolerated both aromatic and aliphatic ketimines, giving the products 31–99% yields with 88:12–99:1 diastereoselectivities and 91–97% ee.
α,β,β-trisubstituted (β2,3,3) amino acids are among the important building blocks for a wide variety of natural products, medicinally relevant molecules, and mimics of protein structural motifs. The ketimine Mannich reaction of α-substituted enolates provides direct access to α,β,β-trisubstituted amino acids from readily available carbonyl compounds. Since they found that the copper enolate generation from a corresponding silicon enolate through transmetalation was the rate-limiting step for their method, they attempted to increase the concentration of active copper enolates by the conjugate addition of Cu-based nucleophiles to α,β-unsaturated esters. This work is not only the first example of but also state of the art for the Mannich reaction between nonactivated ketimines and propionate nucleophiles.
At the outset of their study, they found that the reported reductive method for α,β-unsaturated ketones [32][33] quantitatively reduced dimethylacrylamide in the presence of benzaldehyde, but no aldol products formed, although they fully reproduced the reported reductive aldol reaction. After screening various proton scavengers, they found that activated 4 Å molecular sieves (MS) were optimal. These findings highlighted the exceedingly higher nucleophilicity ofO-trichlorosilyl-N-O-ketene acetal than ketone-derived counterparts that underwent aldol reactions in the absence of proton scavengers. The corresponding α,α-disubstituted ketene acetals lose this electronic benefit due to the allylic strain that pushes their nitrogen lone-pairs out of conjugation with the enol functionality [34][35][36].
In 2008, Tsogoeva and coworkers reported highly enantioselective self-coupling of enamides by using BINOL-derived phosphoric acids (Scheme 31) [37]. They took advantage of an equilibrium between ketimine and enamide in the presence of a chiral acid catalyst to utilizeN-acyl ketimines that are otherwise difficult to employ as electrophiles due to the tautomerization. The method provided the self-coupled products with 15–83% yields with 85–>99% ee. They successfully demonstrated the utility of enantio-enriched self-coupled products by converting one example to a β-aminoketone without essential loss in its optical activity (2).
In 2008, Kumagai, Shibasaki, and coworkers reported a direct catalytic asymmetric addition of allylic cyanides to ketimines (Scheme 32) They became interested in nitriles because these can be viewed as a masked carboxylic acid, are readily available, and could be small enough nucleophiles to overcome the severe steric demands of ketimines. Furthermore, they pointed out that allylic cyanides bear a relatively acidic α-proton (pKa= 21.1 in DMSO), and thus, they could be selectively deprotonated by a base catalyst in the presence of ketimines. To their delight, the reaction scope turned out broad, giving the products in 62–95% yields with <2/98–12/88E/Zratios and 71–94% ee.
They began their study with catalysts made from Bu2Mg and BINAM-derived Schiff bases on the basis of their previous experiences with the aldol reaction of α-methyl-α-isothiocyanato ester and determined that the Schiff base shown inScheme 33was optimal with a model ketimine (Ar = 4-BrC6H6). The reversal of the diastereoselection resulted from the opposite ketimine facial selectivities by the Mg and Sr catalysts. The1H NMR spectra of both catalysts were found to be complicated, which implicated a possibility of the oligomeric structures of theses catalysts. Nonetheless, the Mg provided syn products in 70–99% yields with 90:10–93:7 diastereoselectivities and 80–95% ee, and the Sr catalyst gave anti products in 45–99% yields with 83:17–96:4 diastereoselectivities and 87–97% ee.
In 2013, Kumagai, Shibasaki, and coworkers reported a direct catalytic asymmetric vinylogous ketimine Mannich reaction of γ-butenolides (Scheme 34) For the development of direct catalytic asymmetric carbon–carbon bond-forming transformations under proton-transfer conditions, γ-butenolides have attracted much attention as useful pronucleophiles due to their relatively acidic protons that facilitate the generation of the corresponding dienolates and the high frequency of a γ-butenolide motif present in natural products and biologically relevant compounds. It is notable that prior to their contribution, γ-butenolides had not been employed in direct catalytic asymmetric vinylogous Mannich reactions with ketimines. They successfully demonstrated the importance of soft Lewis acid–soft Lewis base interaction (i.e., Cu···S interaction) by confirming that aN-phosphinoyl ketimine (R1= Ph, R2= Me) barely reacted under the optimized reaction conditions.
In 2014, the groups of Dixon and Nakamura independently reported direct asymmetric Mannich reactions of isocyanoacetates and ketimines (Scheme 35) [38][39]. Despite the importance of α,β-diamino acid building blocks in chemical synthesis of biologically relevant molecules, catalytic enantioselective addition of isocyanoacetates to ketimines were not reported before their contributions.
Dixon and a coworker found that when a combination of cinchona-derived aminophosphine precatalyst38and silver oxide was employed as a binary catalyst system, anti-configured imidazoline products (39) were obtained in 70–98% yields with 73:27–99:1 diastereoselectivities and 90–99% ee (Scheme 35, (1)). In general, excess of either electrophile or nucleophile is necessary for challenging asymmetric catalytic transformations such as the ketimine Mannich reaction. In 2016, they further developed this method to include α-substituted isocyanoacetates whose reaction scope was broad and included various aryl, heteroaryl, and alkyl methyl ketimines (not shown) Hydrolysis of these imidazoline products afforded access to fully substituted α,β-diamino acids in an enantio-enriched form.
Nakamura and coworkers reported that a complex made from alkaloid40and Cu(OTf)2efficiently catalyzed the Mannich reaction of isocyanoacetate in the presence of Cs2CO3(Scheme 35, (2)). Their method provided complementary syn-configured imidazoline products (41) in 45–78% yields with 73:27–99:1 diastereoselectivities and 91–99% ee. Two alkyl methyl ketimines (Ar = PhCH2CH2andiBu in (2)) gave anti diastereomers as major products (anti:syn= 72:28 and 60:40, 54 and 78% yields, 72 and 89% ee, respectively). The enantioselective synthesis of imidazolines with vicinal tetrasubstituted stereocenters was achieved by this new chiral Ni complex.
The method provided vicinal diamines bearing tetra- and tri-substituted contiguous stereogenic centers in 61–99% yields with 77:23–95:5 diastereoselectivities and 83–95% ee. Strangely enough, however, acetophenone-derivedN-(diphenylthiophosphinoyl)ketimine provided only trace amounts of the product under the optimized reaction conditions. They conducted a preliminary mechanistic study and found that the analogousN-diphenylphosphinoyl ketimine (R1= CH2CH2Ph, R2= Me) resulted in much lower conversion and stereoselectivity under the reaction conditions, and this catalytic system exhibited higher performance for differentiating the prochiral face of ketimines rather than that of α-cyano carbanions. As such, the result suggested that the specific activation of the thiophosphinoyl group by a soft-Lewis acidic Cu(I) complex was crucial for the formation of a carbon–carbon bond and efficient stereochemical discrimination.
In 2015, Nakamura and coworkers reported that a complex made from alkaloid42and Zn(OTf)2efficiently catalyzed the ketimine Mannich reaction of a γ-butenolide (Scheme 37) [40]. The ketimine scope was broad and included aryl, heteroaryl, and alkyl methyl ketimines as well as 1-indanone-derived ketimine. It is noteworthy that the pseudo-enantiomeric catalyst gave very similar reactivity and selectivity for three ketimines (R1= 4-MeOC6H4, 4-FC6H4and EtOCOCH2CH2, in 81–84% yields with 93:7–96:4 d.r. and 10:90–8:92 er) in this method, which is not always the case for cinchona-derived catalysts.
3. Activated and/or Cyclic Ketimines in the Allylation Reaction
The addition of allylic organometallic reagents to carbonyl and imine compounds represents an important process in the chemical synthesis (selected reviews; [41][42][43][44]). When ketimines are employed as electrophiles, chiral homoallylic α-tertiary amines are obtained. In light of a double bond in the products that can be viewed as a masked carbonyl, the allylation of the ketimines is analogous to the ketimine Mannich reaction.
In 2012, Lam and coworkers reported the first enantioselective rhodium-catalyzed addition of allylboron reagents to endocyclic ketimines (Scheme 38) [45]. Using only 1.5 mol% of the catalyst complex, 1,2,5-thiadiazolidine-1,1-dioxides (43) underwent the reaction with various allyl-, crotyl- and prenyl-trifluoroborates, affording the corresponding products in 61–89% yields with 17:1–>19:1 diastereoselectivities and 95–99% ee, as did a cyclic sulfamidate imine (45) with allyltrifluoroborate, which provided a corresponding homoallylamine in 83% yield with 93% ee. In subsequent years, they expanded the substrate scope of the method, which included endocyclicN-sulfonyl ketimines bearing CF3andn-butyl groups at the imine carbon atom as well as various potassium allyltrifluoroborates (not shown) [46][47].
In 2018, Yang, Zhang, and coworkers described that the same classes of electrophiles and nucleophiles that were previously studied with chiral rhodium complexes by Lam and coworkers (vide supra) underwent the allylation reaction catalyzed by chiral complexes generated from more cost-effective Co(ClO4)·6H2O and BOX ligands. The method afforded the enantio-enriched homoallylamines in 78–98% yields with 3:1 diastereoselectivity and 53–99% ee (Scheme 39) [48].
Ketimines derived from isatins have attracted much attention as electrophiles because they provide chiral 3-substituted 3-amino-2-oxindoles that are a structural motif found in biologically relevant compounds and natural products, and they are relatively reactive electrophiles, as discussed above. However, asymmetric catalytic allylation of isatin-derived ketimines had not been reported prior to a contribution made by Nakamura and coworkers (Scheme 40) Under the optimized reaction conditions, allylation of ketimines with both electron-donating and -withdrawing substituents provided the corresponding products in 84–96% yields with 82–95% ee.
In 2019, Li and coworkers described the development of an enantioselective asymmetric allylation of isatin-derived ketimines with allylboronates promoted by a binary acid system containing bismuth acetate and chiral phosphoric acid (Scheme 41) [49]. It is notable that most of the ketimines investigated were allylated in less than an hour at room temperature with only 1 mol% of Bi(OAc)3and 2 mol% of chiral phosphoric acid48. The method gave chiral homoallylic α-tertiary amines in 73–99% yields with 85.1:14.9–99.3:0.7 enantiomeric ratios. Regarding the actual nucleophile, they proposed that a chiral allyl bismuth complex generated from allylboronate and two molecules of chiral phosphoric acids through transmetalation would be the allylation species, because the α-selectivity was observed with 1-methylalylbornoic acid pinacol ester, and a positive nonlinear effect between the ees of chiral phosphoric acids and products was observed.
In 2020, Du, Chen, and coworkers reported Cu(I)-catalyzed asymmetricα-allenylation of activated ketimines with 3-butynoates (Scheme 42) Screening of catalysts and reaction conditions was conducted with isatin-derived ketimines. The first two classes of ketimines underwent the reaction at –10 °C and provided the corresponding products after 36 h in 79–97% yields with 77–98% ee, and 64–91% yields with 84–92% ee, respectively. In contrast, isoquinoline-1,2,3-trion-derived ketimines took 72 h at rt to provide the products in 59–63% yields with 73–92% ee.
In 2021, Hu, Xu, and coworkers developed a gold and chiral organocatalyst cooperative catalysis strategy for the allylation of isatin-derived ketimines with readily availableN-propargylamides52(Scheme 43) As they obtained an X-ray crystal structure of the allyl gold intermediate (55, , they hypothesized that allyl gold species resulted through the aromatization of the corresponding vinyl gold species promoted by a basic functionality of squaramide53. On this basis, they proposed that the squaramide electrophilically activated a ketimine via dual-hydrogen bonding, while its nitrogen atom coordinated with the gold catalyst, leading to a formal intramolecular reaction (i.e., bifunctional catalysis by the squaramide).
In 2019, Kürti and coworkers described the first direct catalytic enantioselective allylation of acyclic ketiminoesters to provide α-allyl-α-aryl and α-allyl-α-trifluoromethyl amino esters (Scheme 44) They identified that a complex generated from a commercially available BOX-type ligand (56) and InI as an optimal catalyst for both α-aryl- Their method afforded α-allyl-α-aryl and α-allyl-α-trifluoromethyl amino esters in 85–98% yields and 95–99% ee with 5 mol% catalyst loading in CH2Cl2, and 91–99% yields and 90–99% ee with 10 mol% catalyst loading in THF, respectively. Since an enantiomer of the optimal ligand (56) is not commercially available and would require several steps to synthesize, they found another commercially available ligand57that performed comparably to ligand56with the opposite sense of enantioselection, obtaining the corresponding enantiomers in six α-aryl-
In 2020, Hoveyda and coworkers reported a catalytic regio- and enantio-selective synthesis of trifluoromethyl-substituted homoallylic α-tertiary primary amines (Scheme 45) Unprotected N–H ketimines generated in situ from correspondingN-silyl ketimines were allylated by anO-methyl-L-threonine-derived aminophenol-based boryl catalyst, giving the desired products in 32–91% yields with 45:55–>98:2 α:γ selectivity, 95:5–>98:2Z: Eselectivity, and 88:12–>99:1 enantiomeric ratios. this allylation reaction was demonstrated on a gram-scale with no deterioration of the yield, regio- and stereo-selectivities.
In 2020, Chen and coworkers described an asymmetric allylation of acyclic ketiminoesters through copper-catalyzed carboboronation of allenes (Scheme 46) [50]. They found that the use of a bulkyC2-symmetric NHC was the key to control the chemo-, regio-, diastereo-, and enantio-selectivities in their protocol. While the scope of the electrophiles was limited to alkyl aryl glyoxylate-derived ketiminoesters (Ar1= aryl), that of allenes was found broad, affording the products in 53–96% yields with 4:1–>20:1 diastereoselectivities and 75:25–98:2 enantiomeric ratios.
4. Unmodified Ketimines in the Allylation Reaction
In 2006, Kanai, Shibasaki, and coworkers reported the first catalytic enantioselective ketimine allylation reaction (Scheme 47, (1)) On the basis of their previous studies on the Cu-catalyzed allylboration of ketones, they hypothesized that a highly nucleophilic allylcopper species generated from allylboronate would also be a suitable nucleophile for ketimines. They first investigated the effect of different ketimineN-protecting groups on the reactivity by using achiral CuF·3PPh3as a catalyst and selectedN-benzylketimines for the development of an enantioselective variant. Cyclopentyl-DuPHOS was identified as an optimal chiral ligand for aryl methyl ketimines, providing homoallylic amines in 76–97% yields with 81–93% ee.
With respect to the actual nucleophiles, the chiral NHC-Cu complex produced corresponding chiral (Z)-allyl copper intermediates through borylcupration of monosubstituted allenes with B2(pin)2, which underwent the addition reaction to N–H ketimines. The reaction scope was very broad in terms of both ketimines and allenes, and the primary amine products were obtained in 38–95% yields with 85.5:14.5–>99:1 enantiomeric ratios and exceptional diastereoselectivity ( They noted that reactions of ketimines that contained an α- or β-alkoxy or a benzyl group were inefficient, probably due to facile decomposition (enamine formation and β-elimination, respectively) and that the same applied to trifluoromethyl-substituted ketimines (decomposition to unidentified products). This catalytic method puts forward an expeditious strategy for the synthesis of α-tertiary homoallylamines (β-tertiary-amino carbonyl equivalents) with a very broad substrate scope in high diastereo- and enantio-selectivities, thus providing a solution to an important and persisting problem in catalytic enantioselective synthesis.
In 2019, Yun and coworkers described copper-catalyzed asymmetric intramolecular reductive coupling of (E)-dienyl arenes with a tethered ketimine moiety (Scheme 49) [51]. This transformation was a sequence of a chemo-, regio-, and enantio-selective hydrocupration reaction of (E)-dienes that produced allyl copper species and their subsequent intramolecular addition to ketimines. The reaction scope was broad and provided enantio-enriched 1-benzazepine derivatives bearing two contiguous stereocenters in 11–79% yields with 75:25–100:0 diastereoselectivities and 63–97% ee.
In 2010, Trost and a coworker reported that a zwitterionic complex (Pd-TMM) generated in situ from a Pd catalyst and 1-cyano-2-((trimethylsilyl)methyl)allyl acetate (63) underwent facile cycloaddition reaction with various ketimines (Scheme 50) [52][53]. The electrophile scope was very broad and included aryl alkyl, cyclic, and dialkyl ketimines. Phosphoramidite ligand64was found to be optimal for aryl alkyl and cyclic ketimines and provided the corresponding products in 77–99% yields with 7:1–>20:1 diastereoselectivities and 81–>99% ee but gave an unacceptable level of diastereoselectivity for cyclohexyl methyl ketimine. Ligand65, however, turned out fruitful for dialkyl ketimines, affording the cycloadducts in 50–99% yields with 1:1–>20:1 diastereoselectivities and 84–99% ee.
In 2010, Cramer and a coworker reported one example of enantioselective rhodium(I)-catalyzed intramolecular allylation of a ketimine (Scheme 51, (1)) This transformation was initiated by an imine-directed orthorhodation, followed by a carbometallation of the terminal bond of an allene that produced an allyl rhodium species, which in turn underwent an intramolecular ketimine allylation reaction to give the product where its ester group spontaneously cyclized on to the primary amine moiety formed. In 2013, they extended this protocol to Rh-catalyzed dynamic kinetic resolution of racemic allenes (2) [54]. The substrate scopes of both ketimines and allenes were very broad, affording various cyclic α-tertiary homoallylamines in 38–97% yields with 5:1–>20:1E:Zselectivities and 95:5–99:1 enantiomeric ratios.
In 2014, Peng and Takenaka described one example of enantioselective allylation of an aliphatic ketimine with allyltrichlorosilane catalyzed by their helical-chiral Lewis base catalyst (Scheme 52) [55]. It is often employed for the allylation of aldehydes and aldimines, but its catalytic enantioselective addition to ketimines is not known to the best of our knowledge. Based on their preliminary mechanistic study, they hypothesized that a product (bearing a NSiCl3unit before aqueous work-up) inhibited the catalyst from turning over efficiently in CH2Cl2. A more Lewis basic solvent, THF did improve the catalyst’s turnover but adversely affected the enantioselectivity.
This entry is adapted from the peer-reviewed paper 10.3390/catal11060712
References
- Mannich, C.; Krösche, W. Ueber ein Kondensationsprodukt aus Formaldehyd, Ammoniak und Antipyrin. Arch. Pharm. 1912, 250, 647–667.
- Shamna, S.; Afsina, C.M.A.; Philip, R.M.; Anilkumar, G. Recent advances and prospects in the Zn-catalysed Mannich reaction. RSC Adv. 2021, 11, 9098–9111.
- Hou, X.; Du, D. Recent Advances in Squaramide-Catalyzed Asymmetric Mannich Reactions. Adv. Synth. Catal. 2020, 362, 4487–4512.
- Saranya, S.; Harry, N.A.; Krishnan, K.K.; Anilkumar, G. Recent Developments and Perspectives in the Asymmetric Mannich Reaction. Asian J. Org. Chem. 2018, 7, 613–633.
- Frías, M.; Cieślik, W.; Fraile, A.; Rosado-Abón, A.; Garrido-Castro, A.F.; Yuste, F.; Alemán, J. Development and Application of Asymmetric Organocatalytic Mukaiyama and Vinylogous Mukaiyama-Type Reactions. Chem. A Eur. J. 2018, 24, 10906–10933.
- Kumagai, N.; Shibasaki, M. Recent Advances in Catalytic Asymmetric C–C Bond-Forming Reactions to Ketimines Promoted by Metal-Based Catalysts. Bull. Chem. Soc. Jpn. 2015, 88, 503–517.
- Akiyama, T. 2.16 The Bimolecular and Intramolecular Mannich and Related Reactions. In Comprehensive Organic Synthesis II; Elsevier: Amsterdam, The Netherlands, 2014; Volume 2, pp. 629–681. ISBN 9780080977430.
- Karimi, B.; Enders, D.; Jafari, E. Recent Advances in Metal-Catalyzed Asymmetric Mannich Reactions. Synthesis 2013, 45, 2769–2812.
- Kobayashi, S.; Mori, Y.; Fossey, J.S.; Salter, M.M. Catalytic Enantioselective Formation of C−C Bonds by Addition to Imines and Hydrazones: A Ten-Year Update. Chem. Rev. 2011, 111, 2626–2704.
- Matsunaga, S.; Yoshino, T. Construction of Contiguous Tetrasubstituted Chiral Carbon Stereocenters via Direct Catalytic Asymmetric Aldol and Mannich-Type Reactions. Chem. Rec. 2011, 11, 260–268.
- Shibasaki, M.; Kanai, M. Asymmetric Synthesis of Tertiary Alcohols and α-Tertiary Amines via Cu-Catalyzed C−C Bond Formation to Ketones and Ketimines. Chem. Rev. 2008, 108, 2853–2873.
- Riant, O.; Hannedouche, J. Asymmetric catalysis for the construction of quaternary carbon centres: Nucleophilic addition on ketones and ketimines. Org. Biomol. Chem. 2007, 5, 873.
- Bjørgo, J.; Boyd, D.R.; Watson, C.G.; Jennings, W.B. Equilibrium distribution of E–Z-ketimine isomers. J. Chem. Soc. Perkin Trans. 1974, 2, 757–762.
- Gröger, H. Catalytic Enantioselective Strecker Reactions and Analogous Syntheses. Chem. Rev. 2003, 103, 2795–2828.
- Noble, A.; Anderson, J.C. Nitro-Mannich Reaction. Chem. Rev. 2013, 113, 2887–2939.
- Peng, Q.; Guo, D.; Bie, J.; Wang, J. Catalytic Enantioselective Aza-Benzoin Reactions of Aldehydes with 2H-Azirines. Angew. Chem. Int. Ed. 2018, 57, 3767–3771.
- Sun, L.-H.; Liang, Z.-Q.; Jia, W.-Q.; Ye, S. Enantioselective N-Heterocyclic Carbene Catalyzed Aza-Benzoin Reaction of Enals with Activated Ketimines. Angew. Chem. Int. Ed. 2013, 52, 5803–5806.
- Shen, C.; Wang, R.-Q.; Wei, L.; Wang, Z.-F.; Tao, H.-Y.; Wang, C.-J. Catalytic Asymmetric Umpolung Allylation/2-Aza-Cope Rearrangement for the Construction of α-Tetrasubstituted α-Trifluoromethyl Homoallylic Amines. Org. Lett. 2019, 21, 6940–6945.
- Wang, Y.; Deng, L.-F.; Zhang, X.; Niu, D. Catalytic Asymmetric Synthesis of α-Tetrasubstituted α-Trifluoromethyl Homoallylic Amines by Ir-Catalyzed Umpolung Allylation of Imines. Org. Lett. 2019, 21, 6951–6956.
- Hu, B.; Deng, L. Catalytic Asymmetric Synthesis of Trifluoromethylated γ-Amino Acids through the Umpolung Addition of Trifluoromethyl Imines to Carboxylic Acid Derivatives. Angew. Chem. Int. Ed. 2018, 57, 2233–2237.
- Li, Z.; Hu, B.; Wu, Y.; Fei, C.; Deng, L. Control of chemoselectivity in asymmetric tandem reactions: Direct synthesis of chiral amines bearing nonadjacent stereocenters. Proc. Natl. Acad. Sci. USA 2018, 115, 1730–1735.
- Chen, P.; Zhang, J. Phosphine-Catalyzed Asymmetric Synthesis of α-Quaternary Amine via Umpolung γ-Addition of Ketimines to Allenoates. Org. Lett. 2017, 19, 6550–6553.
- Chen, P.; Yue, Z.; Zhang, J.; Lv, X.; Wang, L.; Zhang, J. Phosphine-Catalyzed Asymmetric Umpolung Addition of Trifluoromethyl Ketimines to Morita-Baylis-Hillman Carbonates. Angew. Chem. Int. Ed. 2016, 55, 13316–13320.
- Wu, Y.; Hu, L.; Li, Z.; Deng, L. Catalytic asymmetric umpolung reactions of imines. Nature 2015, 523, 445–450.
- Curto, J.M.; Dickstein, J.S.; Berritt, S.; Kozlowski, M.C. Asymmetric Synthesis of α-Allyl-α-Aryl α-Amino Acids by Tandem Alkylation/π-Allylation of α-Iminoesters. Org. Lett. 2014, 16, 1948–1951.
- Rostoll-Berenguer, J.; Blay, G.; Pedro, J.R.; Vila, C. Asymmetric Oxidative Mannich Reactions. Adv. Synth. Catal. 2021, 363, 602–628.
- Newhouse, T.; Baran, P.S.; Hoffmann, R.W. The economies of synthesis. Chem. Soc. Rev. 2009, 38, 3010.
- Wender, P.A.; Verma, V.A.; Paxton, T.J.; Pillow, T.H. Function-oriented synthesis, step economy, and drug design. Acc. Chem. Res. 2008, 41, 40–49.
- Trost, B. The atom economy--a search for synthetic efficiency. Science 1991, 254, 1471–1477.
- Suto, Y.; Kanai, M.; Shibasaki, M. Catalytic Enantioselective Mannich-type Reactions of Ketoimines. J. Am. Chem. Soc. 2007, 129, 500–501.
- Hayashi, M.; Sano, M.; Funahashi, Y.; Nakamura, S. Cinchona Alkaloid Amide/Copper(II) Catalyzed Diastereo- and Enantioselective Vinylogous Mannich Reaction of Ketimines with Siloxyfurans. Angew. Chem. Int. Ed. 2013, 52, 5557–5560.
- Sugiura, M.; Sato, N.; Kotani, S.; Nakajima, M. Lewis base-catalyzed conjugate reduction and reductive aldol reaction of α,β-unsaturated ketones using trichlorosilane. Chem. Commun. 2008, 2, 4309.
- Sugiura, M.; Sato, N.; Sonoda, Y.; Kotani, S.; Nakajima, M. Diastereo- and Enantioselective Reductive Aldol Reaction with Trichlorosilane Using Chiral Lewis Bases as Organocatalysts. Chem. Asian J. 2010, 5, 478–481.
- DePorre, Y.C.; Annand, J.R.; Bar, S.; Schindler, C.S. Lewis-Base-Catalyzed Reductive Aldol Reaction To Access Quaternary Carbons. Org. Lett. 2018, 20, 2580–2584.
- Allais, C.; Tsai, A.S.; Nuhant, P.; Roush, W.R. Generation of Stereochemically Defined Tetrasubstituted Enolborinates by 1,4-Hydroboration of α,β-Unsaturated Morpholine Carboxamides with (Diisopinocampheyl)borane. Angew. Chem. Int. Ed. 2013, 52, 12888–12891.
- Nuhant, P.; Allais, C.; Roush, W.R. Diisopinocampheylborane-Mediated Reductive Aldol Reactions: Highly Enantio- and Diastereoselective Synthesis of syn Aldols from N -Acryloylmorpholine. Angew. Chem. Int. Ed. 2013, 52, 8703–8707.
- Baudequin, C.; Zamfir, A.; Tsogoeva, S.B. Highly enantioselective organocatalytic formation of a quaternary carbon center via chiral Brønsted acid catalyzed self-coupling of enamides. Chem. Commun. 2008, 4637–4639.
- Ortín, I.; Dixon, D.J. Direct Catalytic Enantio- and Diastereoselective Mannich Reaction of Isocyanoacetates and Ketimines. Angew. Chem. Int. Ed. 2014, 53, 3462–3465.
- Hayashi, M.; Iwanaga, M.; Shiomi, N.; Nakane, D.; Masuda, H.; Nakamura, S. Direct Asymmetric Mannich-Type Reaction of α-Isocyanoacetates with Ketimines using Cinchona Alkaloid/Copper(II) Catalysts. Angew. Chem. Int. Ed. 2014, 53, 8411–8415.
- Nakamura, S.; Yamaji, R.; Hayashi, M. Direct Enantioselective Vinylogous Mannich Reaction of Ketimines with γ-Butenolide by Using Cinchona Alkaloid Amide/Zinc(II) Catalysts. Chem. A Eur. J. 2015, 21, 9615–9618.
- Talbot, F.J.T.; Dherbassy, Q.; Manna, S.; Shi, C.; Zhang, S.; Howell, G.P.; Perry, G.J.P.; Procter, D.J. Copper-Catalyzed Borylative Couplings with C−N Electrophiles. Angew. Chem. Int. Ed. 2020, 59, 20278–20289.
- Huo, H.-X.; Duvall, J.R.; Huang, M.-Y.; Hong, R. Catalytic asymmetric allylation of carbonyl compounds and imines with allylic boronates. Org. Chem. Front. 2014, 1, 303–320.
- Yus, M.; González-Gómez, J.C.; Foubelo, F. Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev. 2011, 111, 7774–7854.
- Kanai, M.; Wada, R.; Shibuguchi, T.; Shibasaki, M. Cu(I)-catalyzed asymmetric allylation of ketones and ketimines. Pure Appl. Chem. 2008, 80, 1055–1062.
- Luo, Y.; Hepburn, H.B.; Chotsaeng, N.; Lam, H.W. Enantioselective Rhodium-Catalyzed Nucleophilic Allylation of Cyclic Imines with Allylboron Reagents. Angew. Chem. Int. Ed. 2012, 51, 8309–8313.
- Hepburn, H.; Chotsaeng, N.; Luo, Y.; Lam, H. Enantioselective Rhodium-Catalyzed Allylation of Cyclic Imines with Potassium Allyltrifluoroborates. Synthesis 2013, 45, 2649–2661.
- Hepburn, H.B.; Lam, H.W. The Isomerization of Allylrhodium Intermediates in the Rhodium-Catalyzed Nucleophilic Allylation of Cyclic Imines. Angew. Chem. Int. Ed. 2014, 53, 11605–11610.
- Wu, L.; Shao, Q.; Yang, G.; Zhang, W. Cobalt-Catalyzed Asymmetric Allylation of Cyclic Ketimines. Chem. A Eur. J. 2018, 24, 1241–1245.
- Wang, J.; Zhang, Q.; Zhou, B.; Yang, C.; Li, X.; Cheng, J.-P. Bi(III)-Catalyzed Enantioselective Allylation Reactions of Ketimines. Iscience 2019, 16, 511–523.
- Zhao, C.-Y.; Zheng, H.; Ji, D.-W.; Min, X.-T.; Hu, Y.-C.; Chen, Q.-A. Copper-Catalyzed Asymmetric Carboboronation of Allenes to Access α-Quaternary Amino Esters with Adjacent Stereocenters. Cell Rep. Phys. Sci. 2020, 1, 100067.
- Li, D.; Park, Y.; Yoon, W.; Yun, H.; Yun, J. Asymmetric Synthesis of 1-Benzazepine Derivatives via Copper-Catalyzed Intramolecular Reductive Cyclization. Org. Lett. 2019, 21, 9699–9703.
- Trost, B.M.; Stambuli, J.P.; Silverman, S.M.; Schwörer, U. Palladium-Catalyzed Asymmetric [3 + 2] Trimethylenemethane Cycloaddition Reactions. J. Am. Chem. Soc. 2006, 128, 13328–13329.
- Trost, B.M.; Silverman, S.M. Enantioselective Construction of Highly Substituted Pyrrolidines by Palladium-Catalyzed Asymmetric [3+2] Cycloaddition of Trimethylenemethane with Ketimines. J. Am. Chem. Soc. 2010, 132, 8238–8240.
- Tran, D.N.; Cramer, N. Rhodium-Catalyzed Dynamic Kinetic Asymmetric Transformations of Racemic Allenes by the [3+2] Annulation of Aryl Ketimines. Angew. Chem. Int. Ed. 2013, 52, 10630–10634.
- Peng, Z. Development of Helical Chiral Catalysts and Their Application in Asymmetric Catalysis. Ph.D. Thesis, University of Miami, Miami, FL, USA, August 2014.