β-adrenergic receptor stimulation (β-ARS) is a physiological mechanism that regulates cardiovascular function under stress conditions or physical exercise, producing a positive inotropic (enhanced contraction), lusitropic (faster relaxation), and chronotropic (increased heart rate) effect.
- β-adrenergic receptor stimulation
- mathematical modelling
- cardiac electrophysiology
- cardiomyocyte
1. Introduction
β-adrenergic receptor stimulation (β-ARS) is a physiological response mechanism that plays a fundamental role in the regulation of cardiomyocyte activity, producing a positive inotropic (enhanced contraction), lusitropic (faster relaxation), and chronotropic (increased heart rate) effect. Such a multifactorial response is triggered via the activation of the β-adrenergic receptors by the sympathetic nervous system, under either stress conditions or physical exercise, and is also known as the “fight-or-flight” response.
β-adrenergic receptors were first described by Lands et al. in the late 1960s [1,2]. They are situated on the cardiomyocyte membrane and react to different neurotransmitters (norepinephrine, epinephrine) and drugs (isoprenaline). When binding with the adrenergic receptor takes place, it starts a reaction cascade where different cellular substrates become phosphorylated, affecting their individual roles in the overall excitation–contraction coupling. As a result, under healthy physiological conditions, the cardiac action potential shortens, while the intracellular calcium transient exhibits an increased amplitude and a faster decay rate as the main manifestations of β-ARS at the cellular level [3,4]. However, the large number of components involved in the β-adrenergic cascade, and the complexity of these subcellular processes and interactions, make β-ARS signalling highly sensitive to cellular changes and to pathological perturbations. As a result, β-ARS plays a main role in a considerable number of heart diseases [5], and it is well established as an important contributor to cardiomyocyte arrhythmogenicity [6,7,8].
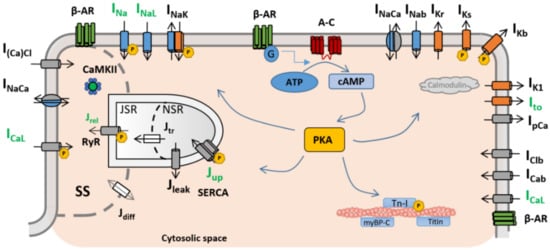
In particular, the β-ARS response has direct effects on the ion channels and pumps of the cell membrane (and, therefore, on intracellular ionic concentrations) regulating calcium intake, intracellular calcium handling, calcium extrusion, and cellular repolarisation. Impairments in the balance between these carefully orchestrated processes can affect heart function and render its constituent cardiomyocytes susceptible to proarrhythmic events, such as early and delayed afterdepolarisations (EADs and DADs, respectively). Such afterdepolarisations under β-ARS are common proarrhythmic manifestations in isolated cardiomyocytes from patients of different pathologies, especially those characterised by action potential and calcium transient abnormalities (such as hypertrophic cardiomyopathy [9], long QT syndrome [10,11], or catecholaminergic polymorphic ventricular tachycardia [12]). An overstimulated β-ARS response is also one of the main mechanisms of cardiac hypertrophy, coronary artery disease, or stroke events [13]. The overexpression of the β-adrenergic response has also been linked to the onset of cardiac hypertrophy or the generation of fibrotic tissue [14,15]. The appearance of these structural changes can lead to the creation of re-entry pathways in myocardial tissue, which also contribute to the generation of self-sustained arrhythmias. Induced arrhythmias are also a common manifestation in heart failure. In chronic heart failure, cardiac remodelling at the structural level can affect the pathways involved in the β-ARS response [16]. As a result, the inotropic response of cardiomyocytes to β-ARS is reduced [17,18], while the propensity to arrhythmogenic events increases. β-adrenergic response is also affected by ageing, and an age-dependent impairment between β-ARS and cardiac function has been demonstrated in both healthy and failing hearts [19,20]. β-ARS is altered as well in severe congenital heart disease patients [21]. Finally, recent studies also suggest a hyperactivation of the positive response of β-ARS patients with coronavirus disease 2019 (COVID-19), potentially leading to life-threatening arrhythmic events [22,23].
Refined knowledge of the role that each cellular component has within the β-ARS cascade, and of the consequences that may arise from disturbing its normal functioning, can therefore lead to a better understanding of different pathologies, as well as to the development of new pharmacological targets for their treatment. In these cases, mathematical modelling and simulation studies of β-ARS can be useful tools for investigating the mechanisms mediating arrhythmic events, assessing their multiscale consequences from the subcellular up to the organ levels, and designing effective treatments [24]. Here, we review the main roles of β-ARS in cellular cardiac electrophysiology, placing our emphasis on the description of the existing mathematical frameworks available for its representation and how insights obtained through experimental approaches have been integrated into these mathematical formulations.
2. Mathematical Models of β-ARS
| Model (Year) | Species | β-ARS Isoform |
Signalling | Substrates | Main Model Advances |
|---|---|---|---|---|---|
| Zeng and Rudy [35] (1995) |
Guinea Pig | Generic | None | ICaL; IK; PLB; INaK; INa | Simulation of the isoproterenol effect by increasing conductances and parameter shift |
| Saucerman et al. [36] (2003) |
Rat | β1 | cAMP; PKA | ICaL; PLB | Dynamic target phosphorylation integrated with cell signalling |
| Greenstein et al. [37] (2004) |
Dog | Generic | None | ICaL; PLB; IKr; IKs | Introduction of a binary population-based approach for target phosphorylation |
| Iancu et al. * [38] (2007) |
Guinea Pig | β1 | cAMP | N/A | Cellular signalling compartmentation |
| Soltis & Saucerman [29] (2010) | Rabbit | β1 | cAMP; PKA | ICaL; IKs; PLB; RyR; TnI; ICFTR | Integration with dynamic CaMKII regulation |
| Hiejman et al. [39] (2011) |
Dog | β1, β2 | cAMP; PKA | ICaL; IKs; IKur; PLB; INaK; INa; RyR; TnI | Two different β isoforms; population-based approach with four different populations |
| Bondarenko [40] (2014) |
Mouse | β1 | cAMP; PKA | ICaL; INa; INaK; RyR; IKur; Ito; IK1; PLB; TnI | Compartmentalised mouse model with new L-type calcium channel subpopulations |
| Khalilimeybodi et al. * [41] (2018) | Mouse | β1, β2 | cAMP; PKA; GSK3β; ERK | N/A | Addition of new molecular signalling pathways (GSK3β and ERK) |
This entry is adapted from the peer-reviewed paper 10.3390/math9151785