SET domain bifurcated 1 (SETDB1) is a histone H3 lysine 9 (H3K9) methyltransferase that exerts important effects on epigenetic gene regulation. SETDB1 complexes (SETDB1-KRAB-KAP1, SETDB1-DNMT3A, SETDB1-PML, SETDB1-ATF7IP-MBD1) play crucial roles in the processes of histone methylation, transcriptional suppression and chromatin remodelling.
- epigenetics
- methyltransferase
- SETDB1
- H3K9
- function
1. Overview
SET domain bifurcated 1 (SETDB1) is a histone H3 lysine 9 (H3K9) methyltransferase that exerts important effects on epigenetic gene regulation. SETDB1 complexes (SETDB1-KRAB-KAP1, SETDB1-DNMT3A, SETDB1-PML, SETDB1-ATF7IP-MBD1) play crucial roles in the processes of histone methylation, transcriptional suppression and chromatin remodelling. Therefore, aberrant trimethylation at H3K9 due to amplification, mutation or deletion of SETDB1 may lead to transcriptional repression of various tumour-suppressing genes and other related genes in cancer cells.
2. Epigenetic Gene Regulation
3. Biological Functions of SETDB1
3.1 The Methylation of Histone H3 Lysine 9 by SETDB1
3.2. Gene transcription silencing by the SETDB1-KRAB-KAP1 complex
The methyltransferase activity of SETDB1 at the H3K9 position in the nuclear euchromatic locus facilitates the combination of the chromo-domain of heterochromatin protein 1α (HP1α) and methylated Lys, which promotes HP1 deposition [9]. HP1 might dimerize with methylated H3K9 in close vicinity and form compacted heterochromatin [46], which contributes to gene silencing. KRAB-associated protein-1 (KAP1), a transcriptional intermediary factor acting as a molecular scaffold among many transcriptional regulatory complexes, was reported to be able to associate with both SETDB1 and HP1α [9]. The connection between SETDB1 and HP1 has been previously confirmed[47]. Specifically, the repression domain of KAP1 binds to the chromo shadow domain of HP1α [48,49]. In addition, KAP1, a common corepressor for the KRAB zinc finger protein (KRAB-ZFP) family (the largest family of sequence-specific DNA binding repressors [50]) of transcriptional repressors [51], connects to KRAB-ZFP through its 75-amino acid KRAB box to form a KRAB-KAP1 repression complex that establishes local microenvironments of heterochromatin at the N-terminal tail of histone H3. As a consequence, the heterochromatin status suppresses gene transcription at specific sites [9,51,52]. Thus, the SETDB1-KRAB-KAP1 repression complex leads to enriched SETDB1, H3K9me3, KRAB, KAP-1, and HP1 at the promoter sequences of specific euchromatic genes, which induces conversion of the target genes from euchromatin to facultative heterochromatin and subsequently represses gene expression [9,53-55].
In mammalian genomes, endogenous retroviruses (ERVs) comprise approximately 8% of the human genome [55]. Although retrotransposition contributes to genome diversification evolution and adaption, it can also lead to genome instability, insertional mutagenesis, or transcriptional perturbation, which is often harmful to host cells [56-59]. ERVs are responsible for 10-12% of the total spontaneous mutations in mice [60]. Aberrant expression of ERVs could alter the expression of neighbouring genes, some of which act as proto-oncogenes to transform host cells [61]. Therefore, multiple defence mechanisms have evolved to maintain the integrity of the genome and transcriptome against retroelement transposition, among which H3K9 methylation modification is important [55] and especially mediated by SETDB1 [34,35,62,63].
ERV silencing is initiated by the recruitment of KAP1 to the target by members of the KRAB-ZFP family. Then, the KAP1/KRAB-ZFP complex is thought to be a pivotal factor for SETDB1 recruitment to retroelements [58]. Simultaneously, HP1 and the NuRD complex are recruited by KAP1 [52]. In detail, KAP1 was discovered to autosumoylate its bromodomain to recruit both the NuRD complex and SETDB1 to the promoter regions of genes modulated by KRAB-ZFPs, which could establish a silent chromatin state by H3K9me3 at genes targeted by KAP1 [9,37]. Recent studies have reported that mouse embryonic stem cells (mESCs) enhance recruitment of SETDB1 to ERV retrotransposition and formation of a KAP1 suppression complex to repress proviral molecules [64]. Furthermore, Peter J. Thompson identified that RNA-binding protein and transcription cofactor heterogeneous nuclear ribonucleoprotein K (hnRNP K) are necessary for the SETDB1-dependent proviral silencing process, which acts as a binding partner of the SETDB1-KAP1 complex by direct interaction in mESCs [65]. Recently, studies of SETDB1 knockout adult mice and differentiated cells demonstrated that SETDB1 also represses retroelements in somatic cells, such as B lymphocytes [66], T lymphocytes [67], neural progenitor cells (NPCs ) [68], and immortalized mouse embryonic fibroblasts (iMEFs) [69]. Mechanistically, SETDB1 is recruited to its target regions by KRAB-ZNF/KAP1 to take effect [70,71], confirming a more general role of SETDB1 in suppressing retroelements.
Recently, SETDB1-dependent ERV repression in cancer cells was also confirmed to be associated with evading recognition by the immune system [72-74]. As a negative regulator of innate immunity, SETDB1 was reported to repress the expression of ERVs through its H3K9-methylating function to preclude the immune response of B cells induced by ERVs, which enables acute myeloid leukaemia (AML) cells to escape innate immunity [72,73]. Above all, the model of SETDB1-KRAB-KAP1 initiated by the recruitment of KAP1 plays a pivotal role in maintaining genome stability and immune regulation, especially through ERVs silencing [74,75].
3.3. Gene transcription silencing by interaction between SETDB1 and DNMT3A
A direct interaction between the N-terminal domain of SETDB1 and the plant homeodomain (PHD) of DNA methyltransferase 3A (DNMT3A) in vivo and in vitro has been shown to facilitate gene transcriptional repression [1]. The repression of the tumour-suppressing gene RASSF1A by a high frequency of methylation at its promoter is widely found in lung, breast, pancreas, kidney, liver, and other cancers [76]. The SETDB1-DNMT3A complex was essential for repressing the promoter of the p53BP2 gene in HeLacells and the RASSF1A gene in MDA-MB-231 breast cancer cells [1,76]. The study also confirmed that SETDB1 is recruited by methyl-CpG DNA Binding Domain Protein 1 (MBD1) to the promoter region of p53BP2 (it seems that MBD1 does not mediate the recruitment of SETDB1 to RASSF1A), and the co-occupation of DNMT3A and SETDB1 leads to a hypermethylated gene promoter [1]. Further study found that SETDB1, DNMT3A, and histone deacetylase 1 (HDAC1) formed a repressive functional complex. During the process, the SETDB1-HDAC1 complex is recruited to the promoter at first to establish trimethylated H3K9 status and represses gene transcription. DNMT3A is later recruited to form theSETDB1-HDAC1-DNMT3A complex and enhance DNA methylation to permanently inactivate the gene. It's a self-propagating epigenetic cycle, in which SETDB1 methylates H3K9 and recruits DNMT3A to reinforce and maintain DNA methylation [1]. Therefore, MBD1-dependent SETDB1-HDAC1 recruitment and SETDB1-mediated H3-K9 methylation initiate the establishment of trimethylated H3K9 status at p53BP2 promoter. SETDB1 recruits DNMT3A which reinforces DNA methylation to connect histone methylation and DNA methylation.
3.4. Gene transcription silencing by interaction between SETDB1 and PML
Promyelocytic leukaemia nuclear bodies (PML-NBs) are large proteinaceous structures that participate in diverse cellular processes such as apoptosis, transcription, cellular senescence, neoangiogenesis, DNA damage response, antiviral response and maintenance of genomic stability, by interacting with multiple proteins [32,33]. Sunwha Cho et al. demonstrated a combination of endogenous SETDB1 and PML proteins from the cleavage stages of development in mice, and their colocalization at PML-NBs was also confirmed. SETDB1 was found to harbor dual functions in this complex. On the one hand, as a necessary part to maintain the structural integrity of PML-NB, SETDB1 physically interacts with the PML protein with its SIM motif [77,78]. On the other hand, SETDB1 acts as a transcriptional regulator of PML-NB-associated genes. The researchers discovered that SETDB1 occupies the promoter of inhibitor of DNA binding 2 (ID2) and methylates H3K9 to prevent ID2 binding to RNA polymerase II, through which it suppresses ID2 expression. This inhibitory progress also depends on the integrity of the PML-NB structure [77,78]. SETDB1-mediated local heterochromatin formation involves a self-reinforcing mechanism: SETDB1-produced H3K9me3 marks recruit HP1 to localize at PML-NB foci [79]. With the aid of HP1, SETDB1 forms a solid platform of heterochromatin to which PML-NB can be riveted [78]. Above all, SETDB1 not only maintains the structure of PML-NB, but also represses the expression of PML-NB-associated genes such as ID2 by trimethylating specific gene locus.
3.5. X chromosome inactivation by SETDB1-ATF7IP-MBD1 complex
X-Chromosome inactivation (XCI), an epigenetic silence caused by heterochromatin formation during the early phase of female mammalian embryonic development, is maintained throughout the lifetime of somatic cells [38]. According to many studies, the accumulation of lncRNA Xist on the X chromatin is the master factor leading to the initiation and maintenance of XCI [80-82]. Interestingly, SETDB1 was also discovered to be related to XCI [38,83]. SETDB1 is the most required methyltransferase to silence approximately 150 genes, which facilitates gene silencing and maintains XCI by alternating the conformation of the whole inactive X chromosome [84]. Minkovsky et al. suggested that during XCI, activating transcription factor 7 interacting protein (ATF7IP or MCAF1), as a bridging factor, interacts with both SETDB1 and the transcriptional repressor domain of MBD1 to form the transcriptional repressor complex SETDB1-ATF7IP-MBD1, which leads to histone H3K9 trimethylation on the inactive X chromosome (Xi) [38,85]. Additional studies confirmed that the formation of the MBD1-chromatin assembly factor-1 (CAF-1) chaperone complex initiates the formation of the transcriptional repressive complex by mediating SETDB1 recruitment to the large subunit of CAF-1 [78,87,88] and maintaining XCI in somatic cells [38,89]. H3K9me3, considered a marker of heterochromatin [77], was enriched at the intergenic, poor gene and repetitive regions of Xi. The heterochromatin structure is inherited during DNA replication through association with MBD1 and ATF7IP [88].
Recently, the location of SETDB1 inside the nucleus was discovered to be mediated by ATF7IP, which increases the ubiquitination of SETDB1 to promote its enzymatic activity [90-92]. The fibronectin type-III (FNIII) domain of ATF7IP plays a certain role in the transcriptional repression function mediated by the SETDB1-ATF7IP complex. However, the FNIII domain seems to be unrelated to the nuclear localization of SETDB1 and to the ATF7IP-dependent integrated retroviral transgenes silencing [92]. Above all, by alternating the conformation of X chromosome through SETDB1-ATF7IP-MBD1 complex, SETDB1 also plays an important role in XCI.
3.6. Remodelling of chromatin associated with SETDB1 expression
In protein posttranscriptional modification, histone methyltransferases regulate the folding and remodelling of chromatin by modifying the N-tail of histones [42]. SETDB1 is a histone H3K9-specific methyltransferase that associates with various transcription factors to regulate gene expression via chromatin remodelling, reflecting that SETDB1 is closely related to chromatin modification and heterochromatin formation [20,93,94]. Recently, some studies reported that the master silencing factor KAP1 recruits other factors to establish interstitial heterochromatin in mESCs, especially SETDB1 [55,94]. During the formation of the SETDB1-KRAB-KAP1 repressive complex mentioned above, the interaction between HP1α and KAP1 triggers the conversion of target foci from euchromatin to heterochromatin, thereby inhibiting gene expression [9,51]. H3K9 methyltransferases have also been confirmed to be the "gatekeepers" of chromatin condensation. Methylated H3K9 can recruit H1 linker histones and HP1, while the localization of histone H1 is associated with the gliding of nucleosomes [95]. In addition, SETDB1 can also combine with DNMTs or HDACs to remodel the chromatin structure [96]. Many studies have confirmed that SETDB1 plays an essential role in chromatin remodelling, especially heterochromatin formation, through interacting with other factors and its H3K9 methylation function.
3.7. Early embryo development associated with SETDB1
In mouse embryos, SETDB1 holds a prominent position in early development. SETDB1 dysfunction has been found to induce the earliest lethality around peri-implantation compared with other H3K9-specific HKMTs such as SUV39h1 (nonessential) [97], G9a (die around day 9.5) [98], and GLP (die around day 9.5) [99]. Cho et al. found that SETDB1 discontinuously appears in the pronucleus after fertilization, and its expression in males is higher than its expression in females [100,101], indicating that SETDB1 may participate in the restructuring of sperm-derived chromatin [22,102]. The expression of SETDB1 turns to a diffuse pattern until the 2-cell stage [102] but temporarily fades in the 8-cell stage before reappearing in a spotted form again in the blastocyst [77,103]. During the blastocyst phase, restored SETDB1 localizes to PML-NB foci [103] and is expressed equally in the inner cell mass (ICM) and trophectoderm (TE) cells.SETDB1 is then expressed only in the ICM during the blastocyst outgrowth phase [102]. The varying expression patterns of SETDB1 during the preimplantation stage suggest crucial functions of SETDB1 in mouse early embryonic development. The catalytic activity of SETDB1 is necessary in meiosis and early oogenesis, without it, a decrease in the number of mature eggs as a result [104,105].
3.8. Embryonic stem cell development associated with SETDB1
Blastocysts comprise ICM and TE cells. The punctate pattern of SETDB1, as mentioned in 2.7., was only expressed in ICM-derived Oct4-positive cells during the blastocyst outgrowth phase. Furthermore, ESCs can be established in vitro by extended culture of ICM cells [78], which is consistent with the result that SETDB1-null blastocysts fail to give rise to ESCs in vitro [106], suggesting the pivotal role of SETDB1 in the establishment and/or maintenance of ESCs.
SETDB1 was found to maintain the pluripotency of mESCs by repressing differentiation-associated genes [78] and trophoblastic genes [107]. Conditional knockout of SETDB1 in mESCs induced the expression of trophectoderm differentiation markers such as caudal type homeobox 2 (Cdx2), transcription factor AP-2 alpha (Tcfap2a), and heart and neural crest derivatives expressed 1 (Hand1) [108]. Additional data found that SETDB1 interacts with Oct4, which in turn recruits SETDB1 to silence trophoblast-associated genes. These findings demonstrate that SETDB1 restricts the extraembryonic trophoblast lineage potential of pluripotent cells [108]. Recently, a study demonstrated that SETDB1 restricts the transition from pluripotency to totipotency in ESCs by silencing ERV and relevant genes [109]. SETDB1 is also essential to mESC self-renewal [107,110,111]. Lack of SETDB1 resulted in downregulation of Nanog, SRY-box 2 (Sox2) and POU class 5 homeobox 1 (Pou5f1) genes associated with induced pluripotent stem cell (iPSC) reprogramming and positive regulation of pluripotency and self-renewal of mESCs [108].
A recent study demonstrated that the H3K9 methylation reader M-phase phosphoprotein 8 (MPP8) cooperates with SETDB1 physically and functionally to coregulate a great number of common genomic targets, especially the DNA satellite, which silences satellite DNA repeats in mESCs [85].
4. Brief summary of the functions of SETDB1
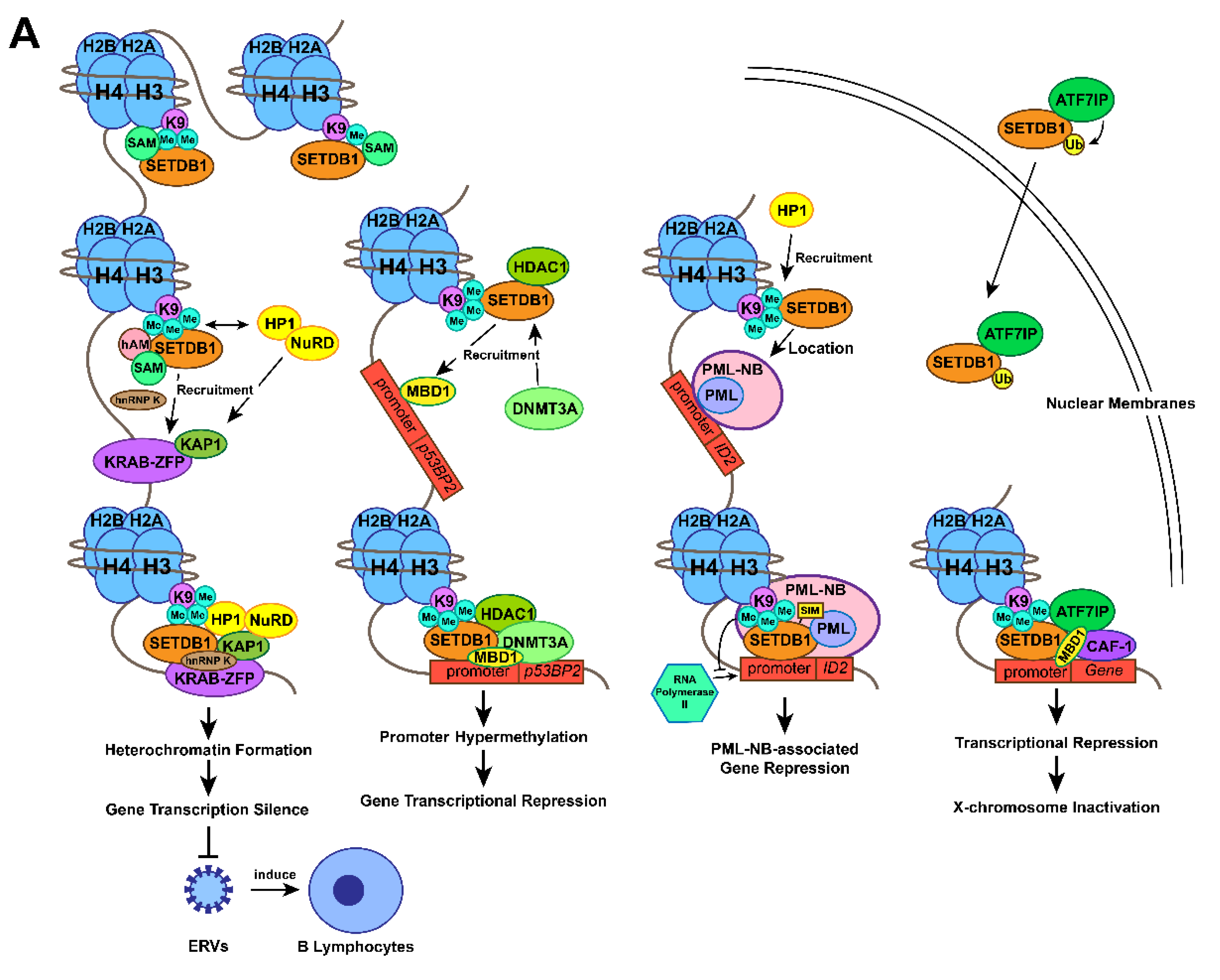
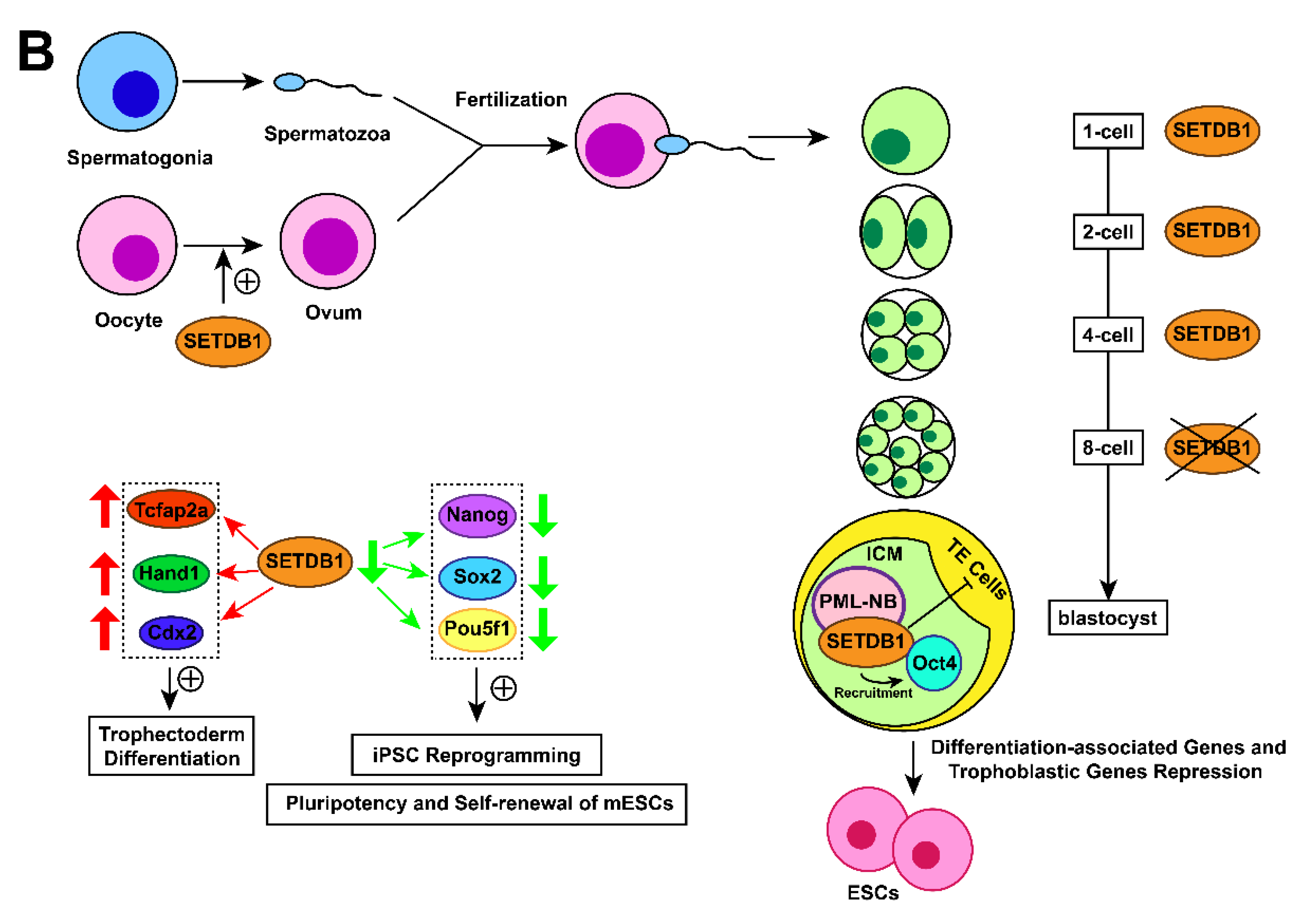
Figure 1. Action mechanisms and the subsequent functions of SETDB1 with its cofactors. (A) Together with H3K9 trimethylation on specific gene locus, the formation of SETDB1-KRAB-KAP1, SETDB1-DNMT3A, SETDB1-PML, SETDB1-ATF7IP-MBD1 complexes play crucial roles in the processes of histone methylation, chromatin remodelling and transcriptional suppression, which leads to corresponding physiological functions, including ERVs silencing, XCI and tumour-suppressive genes repression. (B) SETDB1 exerts different roles during the preimplantation stage due to its varying expression patterns. It maintains the pluripotency of mESCs by repressing differentiation-associated and trophoblastic genes while facilitating self-renewal by positively regulating the genes associated with iPSC reprogramming and self-renewal.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22147416
References
- Li, H.; Rauch, T.; Chen, Z.X.; Szabó, P.E.; Riggs, A.D.; Pfeifer, G.P. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 2006, 281, 19489–19500.
- Cruz-Tapias, P.; Zakharova, V.; Perez-Fernandez, O.M.; Mantilla, W.; RamÍRez-Clavijo, S.; Ait-Si-Ali, S. Expression of the Major and Pro-Oncogenic H3K9 Lysine Methyltransferase SETDB1 in Non-Small Cell Lung Cancer. Cancers 2019, 11, 1134.
- Jambhekar, A.; Dhall, A.; Shi, Y. Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell Biol. 2019, 20, 625–641.
- Yu, C.; Zhuang, S. Histone Methyltransferases as Therapeutic Targets for Kidney Diseases. Front. Pharmacol. 2019, 10, 1393.
- Bedford, M.T.; Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272.
- Hamamoto, R.; Saloura, V.; Nakamura, Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 2015, 15, 110–124.
- McGrath, J.; Trojer, P. Targeting histone lysine methylation in cancer. Pharmacol. Ther. 2015, 150, 1–22.
- Saha, N.; Muntean, A.G. Insight into the multi-faceted role of the SUV family of H3K9 methyltransferases in carcinogenesis and cancer progression. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188498.
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., 3rd. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932.
- Wang, H.; An, W.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Chatton, B.; Tempst, P.; Roeder, R.G.; Zhang, Y. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol. Cell 2003, 12, 475–487.
- Sun, Q.Y.; Ding, L.W.; Xiao, J.F.; Chien, W.; Lim, S.L.; Hattori, N.; Goodglick, L.; Chia, D.; Mah, V.; Alavi, M.; et al. SETDB1 accelerates tumourigenesis by regulating the WNT signalling pathway. J. Pathol. 2015, 235, 559–570.
- Rodriguez-Paredes, M.; Martinez de Paz, A.; Simó-Riudalbas, L.; Sayols, S.; Moutinho, C.; Moran, S.; Villanueva, A.; Vázquez-Cedeira, M.; Lazo, P.A.; Carneiro, F.; et al. Gene amplification of the histone methyltransferase SETDB1 contributes to human lung tumorigenesis. Oncogene 2014, 33, 2807–2813.
- Wang, G.; Long, J.; Gao, Y.; Zhang, W.; Han, F.; Xu, C.; Sun, L.; Yang, S.C.; Lan, J.; Hou, Z.; et al. SETDB1-mediated methylation of Akt promotes its K63-linked ubiquitination and activation leading to tumorigenesis. Nat. Cell Biol. 2019, 21, 214–225.
- Chen, B.; Wang, J.; Wang, J.; Wang, H.; Gu, X.; Tang, L.; Feng, X. A regulatory circuitry comprising TP53, miR-29 family, and SETDB1 in non-small cell lung cancer. Biosci. Rep. 2018, 38.
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416.
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565.
- Kang, H.C.; Kim, H.K.; Lee, S.; Mendez, P.; Kim, J.W.; Woodard, G.; Yoon, J.H.; Jen, K.Y.; Fang, L.T.; Jones, K.; et al. Whole exome and targeted deep sequencing identify genome-wide allelic loss and frequent SETDB1 mutations in malignant pleural mesotheliomas. Oncotarget 2016, 7, 8321–8331.
- Yoshikawa, Y.; Emi, M.; Nakano, T.; Gaudino, G. Mesothelioma developing in carriers of inherited genetic mutations. Transl. Lung Cancer Res. 2020, 9, S67–S76.
- Torricelli, F.; Lococo, F.; Di Stefano, T.S.; Lorenzini, E.; Piana, S.; Valli, R.; Rena, O.; Veronesi, G.; Billè, A.; Ciarrocchi, A. Deep Sequencing Analysis Identified a Specific Subset of Mutations Distinctive of Biphasic Malignant Pleural Mesothelioma. Cancers 2020, 12, 2454.
- Karanth, A.V.; Maniswami, R.R.; Prashanth, S.; Govindaraj, H.; Padmavathy, R.; Jegatheesan, S.K.; Mullangi, R.; Rajagopal, S. Emerging role of SETDB1 as a therapeutic target. Expert Opin. Ther. Targets 2017, 21, 319–331.
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential. Cancer Res. 2021, 81, 525–534.
- Yu, L.; Ye, F.; Li, Y.Y.; Zhan, Y.Z.; Liu, Y.; Yan, H.M.; Fang, Y.; Xie, Y.W.; Zhang, F.J.; Chen, L.H.; et al. Histone methyltransferase SETDB1 promotes colorectal cancer proliferation through the STAT1-CCND1/CDK6 axis. Carcinogenesis 2020, 41, 678–688.
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016.
- de Thé, H.; Le Bras, M.; Lallemand-Breitenbach, V. The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21.
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell. Mol. Life Sci. 2017, 74, 2055–2065.
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16.
- Murry, C.E.; Keller, G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 2008, 132, 661–680.
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931.
- Minkovsky, A.; Sahakyan, A.; Rankin-Gee, E.; Bonora, G.; Patel, S.; Plath, K. The Mbd1-Atf7ip-Setdb1 pathway contributes to the maintenance of X chromosome inactivation. Epigenet. Chromatin 2014, 7, 12.
- Li, F.; Ye, B.; Hong, L.; Xu, H.; Fishbein, M.C. Epigenetic modifications of histone h4 in lung neuroendocrine tumors. Appl. Immunohistochem. Mol. Morphol. AIMM 2011, 19, 389–394.
- Fischle, W.; Wang, Y.; Allis, C.D. Histone and chromatin cross-talk. Curr. Opin. Cell Biol. 2003, 15, 172–183.
- Herz, H.M.; Garruss, A.; Shilatifard, A. SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 2013, 38, 621–639.
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837.