The reinforced link between diabetes and cancer and/or breast cancer has generated interest in studying the effects of widely prescribed anti-hyperglycemic/anti-diabetic drugs on the risk, incidence, progression, response to therapy, resistance, and post-therapeutic relapse in cancers. The incidence and rising rates of diabetes is a serious concern in the medical field around the globe. Worldwide, the World Health Organization (WHO) data estimate nearly 422 million diabetes affected individuals in 2014, a sharp rise when compared to the 108 million diabetes affected individuals in 1980 [
1]. Data projections suggest that the global diabetes prevalence of 8.8% in 2017 will further increase to 9.9% by 2045 [
2]. While it is well established that diabetes is linked to a higher risk of cardiovascular diseases, hepatic and renal complications, and nerve damage, much less appreciated is the fact that diabetes can be linked to a higher risk, incidence, progression, and post-treatment prognosis of different cancers [
3,
4,
5,
6,
7]. More recently, owing to the many common risk factors attributable to both diabetes and cancer a convincing link was established, by several epidemiological studies, between the occurrence of diabetes and the higher risk and incidence of many different cancers, including the various types of breast cancer [
5,
8,
9,
10,
11,
12]. In this regard, particularly in breast cancers, while insulin and insulin analogues used to treat diabetes propagated tumor growth through the induction of angiogenesis and activation of mitogenic signaling mechanisms and drugs such as thiazolidinediones do not appear to have a significant anti-cancer effect, metformin on the other hand exhibited significant anti-proliferative and anti-cancer effects [
5].
Metformin (1, 1-dimethylbiguanide) has its history traced back to the 18th century (year 1772), when
Galega officinalis (commonly known as French Lilac/Goat’s Rue/Spanish Safonin/False Indigo) was used to treat symptoms which was later attributed to diabetes [
13,
14]. While the hypoglycemic activity of
Galega officinalis was attributed to the guanidine component by the 1800s, the apparent toxicity associated with the clinical use of guanidine led to synthesis, testing, and use of several biguanides, including dimethylbiguanide, for their glucose-lowering and anti-malarial effects and for the treatment of influenza in the late 1920s [
13,
14]. It was then in 1957 that Dr. Jean Sterne published his studies on metformin and proposed its clinical development and the name ‘Glucophage’ (meaning glucose-eater) for metformin [
13,
14]. Metformin was thrust into the limelight as a better anti-hyperglycemic drug by the late 1970s, when its cousins, the biguanides such as phenformin and buformin (which had more potent glucose-lowering effect), were associated with lactic acidosis and had to be discontinued in medicinal practice [
13,
14]. Metformin on the other hand reportedly has only mild to moderate side effects such as nausea, vomiting, and diarrhea, which can be rectified by treatment dosage adjustments [
15]. However, predominantly in elderly individuals, with heart failure, hypoxia, sepsis, renal and hepatic comorbidities, and dehydration, metformin administration can lead to lactic acidosis in rare cases [
15,
16,
17,
18]. The confirmed anti-hyperglycemic effect (without causing hypoglycemia) and the favorable safety prolife when compared to phenformin and buformin helped metformin claim the title as the ‘most widely prescribed and first-line oral anti-diabetic drug’ and manages to keep that title 62 years after its first clinical use in the treatment and management of type 2 diabetes [
13,
14,
19].
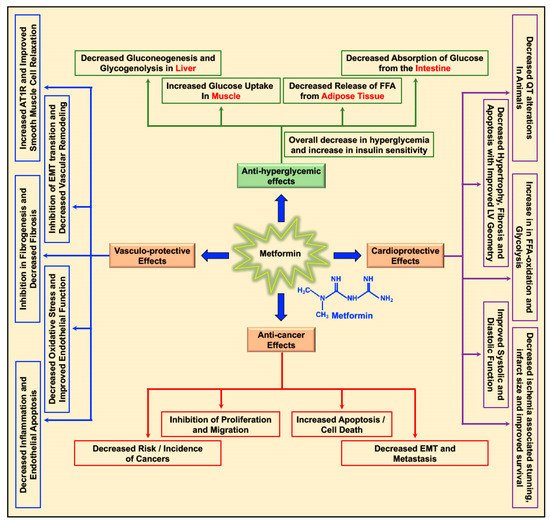
Metformin decreases the levels of blood glucose by decreasing gluconeogenesis and glycogenolysis in the liver, decreasing the intestinal absorption of glucose, reducing the release of free fatty acids (FFA) from adipose tissue, and increasing glucose utilization by the muscle (
Figure 1) [
20]. Apart from its glucose-lowering effect, metformin was studied for its cardioprotective and vasculo-protective effects and more recently for its effects as a cancer preventive and anti-cancer/anti-tumor agent in different cancers (
Figure 1) [
5,
20,
21]. Depending on patient prolife and various disease conditions or stages, metformin treatment-associated beneficial effects in the treatment of hepatic diseases [
22,
23,
24,
25], renal damage and disorders [
26], neurodegenerative diseases [
27,
28,
29], and bone disorders [
30] were reported. In addition, metformin treatment-related antiaging effects, delay in the onset of age-related disorders, and improvement in longevity (lifespan) were reported in
C. elegans, insects, and rodents [
31,
32,
33,
34].
2. Biology of Metformin and Molecular Mechanism of Action
In a type 2 diabetic individual who receives metformin orally, the concentration of metformin in the hepatic circulation may reach 50 μM; with the peak plasma concentration of metformin at 20 μM [
5,
48,
49]. The hydrophilic and cationic nature of metformin at physiological pH makes it highly unlikely that metformin rapidly diffuses through the cell membrane and exerts it effect on cell function. In addition, the kidneys carry out the elimination of unaltered metformin through the urine [
50]. Hence, it is evident that metformin requires the presence and support of transporter molecules for its absorption, distribution, and elimination to exert its biological function. In this regard, the organic cation transporters 1, 2, and 3 (OCT1, OCT2, and OCT3), the plasma membrane monoamine transporter (PMAT), and multidrug and toxin extrusion protein 1 and 2 (MATE1 and MATE2) transporters are reported to play key roles in transporting metformin into and out of the cell in the intestine, liver, and kidney [
50,
51,
52,
53,
54,
55,
56,
57]. The thiamine transporter 2 (THTR2) also plays a role in intestinal absorption and renal re-absorption of metformin [
58]. Alterations in the OCT1 gene reduced hepatic uptake of metformin and reduced the efficacy of metformin in reducing blood glucose levels by the inhibiting gluconeogenesis and glycogenolysis [
59,
60].
While several studies have reported various ‘AMPK dependent’ and ‘AMPK independent’ mechanisms for the anti-cancer/anti-tumor effects of metformin in cancer therapy, these anti-cancer effects of metformin were only observed at very high concentrations (>5 mM) and fall short of explaining how such high concentrations enters the cancer cells and exerts its anti-neoplastic effect. Studies have implicated that the susceptibility and/or resistance of cancer cells to metformin treatment is dependent on the varying levels of the cell surface metformin transporters. Overexpression of OCTs that contribute to intracellular accumulation of metformin in cancers would make them susceptible to metformin treatment (which should explain the high concentrations of metformin required for anti-cancer treatment) while the overexpression of MATE transporters that contribute to the extrusion of metformin out of the cell would render the cancer cell resistant to metformin treatment (
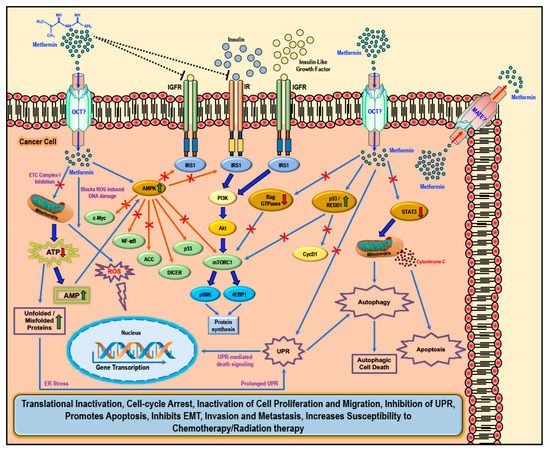
Figure 2) [
61,
62,
63,
64].
Figure 2. Cellular anti-cancer/anti-tumor effects of metformin: The hydrophilic and cationic metformin is transported into the cell via the organic cation transporters (OCT), which support the intracellular accumulation of metformin. The anti-proliferative activity of metformin in several cancers is at least in part attributed to its ability to reduce the levels of insulin/IGF1, which in turn inhibits the insulin/IGF1 mediated molecular pathways that support tumor initiation and progression. Metformin treatment directly activates AMPK and the ‘AMPK dependent’ effects include inhibition of c-Myc, NF-κB, and mammalian target of rapamycin-C1 (mTORC1) pathways and acetyl Co-A carboxylase (ACC)-dependent lipogenesis pathways while activating the p53 pathway and DICER-mediated miRNA synthesis. Metformin, albeit at high concentrations, is also known to inhibit the mitochondrial Complex 1 of the electron transport chain (ETC) thereby reducing ATP, levels which increases the AMP/ATP ratio further leading to AMPK activation. A decrease in ATP/energy levels can also lead to mismanaged protein folding mechanisms leading to the accumulation of unfolded or misfolded proteins and prolonged unfolded protein response (UPR) without rectification of endoplasmic stress triggers apoptosis through multiple mechanisms, which include activation of UPR mediated apoptotic/death signaling and activation of autophagy and subsequent autophagic cell death. AMPK independent metformin treatment-associated anti-cancer effects are mediated by Rag GTPases, REDD1, and STAT3. Overall metformin treatment in cancer cells causes translational inactivation, cell-cycle arrest, inactivation of cell proliferation and migration, inhibition of UPR, promotes apoptosis, inhibits EMT, invasion, and metastasis, and increases susceptibility to chemotherapy/radiation therapy.
There are two general mechanisms that could explain the putative anti-cancer effects of metformin. The ‘indirect’ anti-cancer effects of metformin arise from its ability to reduce insulin resistance, insulin levels, and fasting glucose levels [
65]. Physiologically, insulin and insulin-like growth factor-1 (IGF1) largely regulate carbohydrate and lipid metabolism and storage and protein synthesis via transmembrane receptor binding and activation of receptor tyrosine kinase and subsequent activation of intracellular insulin receptor substrate-1 (IRS1); however, insulin and IGF1-mediated signaling pathways are also implicated in pathogenesis and progression of several cancers via the activation of the Ras/Raf/MEK/ERK, PI3K/Akt/mTORC1, and GSK3β/β-catenin pathways [
5,
44]. Metformin reduces blood glucose levels in circulation by decreasing gluconeogenesis and glycogenolysis in the liver, decreasing the intestinal absorption of glucose, reducing the release of FFA from adipose tissue, and increasing glucose utilization by the muscle. Lower blood glucose concentration in turn decreases the synthesis and secretion of insulin by the β-cells of the pancreas and reduces the levels of insulin in circulation. The anti-proliferative activity of metformin in several cancers is at least in part attributed to its ability to reduce the levels of insulin/IGF1, which in turn inhibits the insulin/IGF1 mediated molecular pathways that support tumor initiation and progression rather than a direct anti-proliferative/anti-cancer action (
Figure 2) [
44].
Metformin also exhibited ‘direct’ anti-cancer effects in many different cancer related studies. Since cancer cells are known to utilize glucose rapidly through glycolysis (Warburg effect) to meet their energy needs when compared to normal cells, metformin-mediated decrease in glucose levels should also curb tumor growth, although reports suggest that cancer cells use alternative sources of energy when starved of glucose or when glycolysis is inhibited [
45,
66,
67,
68]. Furthermore, AMPK inhibits IRS1 mediated IR and IGFR oncogenic signaling via PI3K/Akt/mTOR, which, potentially, also contributes to the anti-cancer effect of metformin (
Figure 2) [
69,
70,
71].
Several experimental studies reported the ‘direct’ anti-cancer effects of metformin, which are distinct from its ‘indirect’ anti-cancer effects that are related to its anti-hyperglycemic actions, inhibition of hepatic gluconeogenesis, and reduction of insulin signaling [
72,
73,
74]. In cancer cells, aberrant signaling mechanisms were reported involving key proteins and their related pathways associated with protein synthesis and survival (mTOR, c-Myc, and NF-κB), lipid synthesis (ACC), DNA damage repair and apoptosis (p53), and miRNA synthesis and function (DICER). Each of these proteins and their modulation/regulation can impact the incidence, growth, and progression of malignant tumors. Mammalian target of rapamycin-C1 (mTORC1) is activated and upregulated by nutrients, growth factors and energy and stress signals, and key signaling pathways (PI3K, MAPK, and AMPK) and is inhibited by rapamycin [
75,
76,
77]. Activation of mTORC1 regulates cellular protein synthesis and cell survival through the phosphorylation of its substrates, 4EBP1, and p70S6K [
75,
76,
77]. The central role of mTOR in regulating cellular protein synthesis and cell survival explains the association of an overactive mTOR pathway cancer [
75,
76,
77]. Several mTOR inhibitors were successfully tested for the treatment of various cancers [
75,
76,
77]. Cellular Myc (c-Myc) is a well-studied oncogene which was constitutively overexpressed in various cancers [
78,
79]. Myc reportedly regulates an array of cellular functions that support cancer cell growth and progression including transcription of several oncogenes, translation, cell cycle progression, cell proliferation, differentiation and survival, ribosome biogenesis, signal transduction, and also cancer stem cell-related signaling and resistance to cancer treatment [
78,
79,
80]. NF-κB activation, similarly, promotes cancer growth and by regulating transcription of genes that support cell proliferation and survival, angiogenesis, tumor progression, and metastasis [
81,
82,
83]. The DNA repair and pro-apoptotic nuclear transcription factor, p53, is vital to tumor suppression [
84]. Loss of p53 function and mutations in the p53 gene are notably the cause for the incidence and progression of many different cancers by supporting cell proliferation and survival, metabolism, genome instability, pro-survival autophagy, and metastasis in addition to conferring therapeutic resistance to cancer cells [
84,
85,
86]. The RNase enzyme, DICER, that is important in processing the formation of function micro-RNAs (miRNA) was frequently downregulated in human cancers and was linked to cancer progression and cancer metastasis [
87,
88,
89].
Metformin-mediated activation of AMPK and subsequent modulation and regulation of intracellular proteins and their functions can explain several of the biological functions as well as its anti-cancer/anti-proliferative effects that was observed in most cancer cells (
Figure 2) [
44,
45,
90,
91,
92,
93]. Activation of AMPK in cancer cells is associated with inhibition of the mTORC1, c-Myc, and NF-κB pathways and activation of DICER and the p53 pathway, all of which reportedly exert tumor suppressive, anti-proliferative, anti-migratory, and pro-apoptotic effects through various intracellular mediators, activation of anti-oncogenic genes, and downregulation of pro-oncogenic genes [
94,
95,
96,
97,
98,
99,
100,
101,
102,
103,
104,
105]. Metformin treatment-associated AMPK activation leads to the phosphorylation of tuberous sclerosis-2 (TSC2) or raptor and subsequent mTORC1 pathway inhibition, thereby reducing the cellular translational process/protein synthesis and overall cell survival [
93,
106,
107,
108,
109,
110]. AMPK also phosphorylates and inhibits acetyl CoA carboxylase (ACC), thereby reducing lipid biosynthesis (
Figure 2) [
111]. Inhibition of these anabolic processes of protein and lipid biosynthesis thus retards cancer cell growth and proliferation [
112].
Furthermore, metformin can inhibit the mitochondrial respiratory chain complex 1 (
Figure 2), thereby causing a reduction in the NADH oxidation and the proton gradient across the inner mitochondrial membrane subsequently reducing the rate of oxygen consumption [
113,
114]. Since cancer cells are highly glycolytic in nature (Warburg effect) and depend less on the oxidative phosphorylation for its energy needs (ATP), it can be argued that the effect of metformin as an inhibitor of the electron transport chain (ETC) complex 1 may be weak and reversible and may not impact the growth or proliferation of cancer cells [
47]. However, any cancer cell that utilizes oxygen for mitochondrial respiration would produce mitochondrial ATP and, thus, a decrease in ATP production due to metformin-mediated inhibition of ETC complex 1 should be toxic to the cells [
47]. Additionally, the increasing levels of AMP due to ETC complex 1 inhibition should also in turn activate AMPK (
Figure 2) [
46,
115]. The decrease in ATP also contributes to the accumulation of unfolded/misfolded proteins [
116,
117,
118]. The accumulation of misfolded or unfolded proteins turns on the endoplasmic reticulum stress/unfolded protein response (UPR) pathway [
119]. Prolonged UPR and accumulation of unfolded proteins without rectification of endoplasmic reticulum stress triggers apoptosis through multiple mechanisms, which include activation of UPR-mediated apoptotic/death signaling and activation of autophagy and subsequent autophagic cell death [
119]. Additionally, the ER stress mediated release of calcium (Ca
2+) from the endoplasmic reticulum stores leads to Ca
2+ accumulation in the mitochondria causing depolarization of permeability transition pore (PTP) and inducing apoptosis via the release of caspases [
119].
Metformin treatment-associated ‘AMPK independent’ anti-cancer effects are mediated by regulated in DNA Damage-1 (REDD1; also known as DNA damage inducible transcript-4-DDIT4), Rag GTPases, and signal transducer and activator of transcription-3 (STAT3) (
Figure 2). REDD1/DDIT4 is known to be inhibitor of mTOR signaling and thereby possess tumor suppressive properties by inhibition of protein synthesis and cell survival [
120,
121,
122,
123]. Metformin reportedly activated the p53/REDD1 axis to cause AMPK independent inhibition of mTOR in cancer cells (
Figure 2) [
124]. Activation of the p53/REDD1 pathway also reduces the expression of Cyclin D1, thereby reducing cell proliferation [
125]. Rag GTPases, a sub-family of Ras-related GTPases, is involved in amino acid signaling mediated activation and functioning of the mTOR pathway [
126,
127,
128]. Metformin treatment inhibits the mTOR pathway via the inhibition of Rag GTPases in cancer cells, which in turn reduces protein synthesis and causes cell cycle arrest (
Figure 2) [
125,
129,
130]. The aberrant activity of STAT3 has been implicated in promoting the pro-oncogenic functions such as initiation, progression, metastasis, and immune evasion in different cancers [
131,
132]. Overexpression of STAT3 contributes to cell survival, proliferation, cell cycle progression, anti-apoptosis, migration, invasion, angiogenesis, chemoresistance, immunosuppression, and self-renewal and differentiation of stem cells by regulating the expression of its downstream target genes [
131,
132]. Metformin treatment inhibited STAT3 nuclear translocation and exerted anti-proliferative, anti-metastatic, and pro-apoptotic effects in cholangiocarcinoma cells and breast cancer cells (
Figure 2) [
133,
134].
The multifaceted ability of metformin to influence cancer cell growth and cancer progression through various molecular mechanisms, as discussed above, has made it an interesting candidate drug with potential in the treatment of breast cancer. In the following sections of the article, we briefly discuss the cellular, pre-clinical, and clinical studies that are currently testing metformin as a monotherapy or in combination with other chemotherapeutic drugs or phytochemicals/natural compounds for its efficacy as an anti-cancer/anti-tumor agent in the treatment of estrogen receptor (ER) positive, progesterone receptor (PR) positive, human epidermal growth factor receptor 2 (HER2) positive types of breast cancers, and triple negative breast cancers (TNBCs) [
135].