The surface chemistry of carbon materials is predominantly explored using x-ray photoelectron spectroscopy (XPS). However, many journal articles have critical failures in the published analysis which typically stems from an ill-informed approach to analyzing the spectroscopic data. The presented work presents a discussion on lineshapes and associated changes in the spectral envelope of predominantly graphitic materials, which together with the use of the D-parameter to verify levels of the graphitic content, using this information to highlight a simple and logical approach to strengthen confidence in the functionalization derived from the carbon core-level spectra.
- carbon
- XPS
- oxygen
- fitting
- quantification
1. Introduction
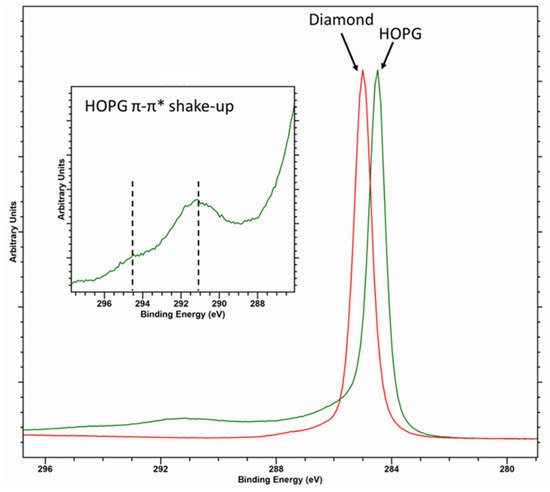
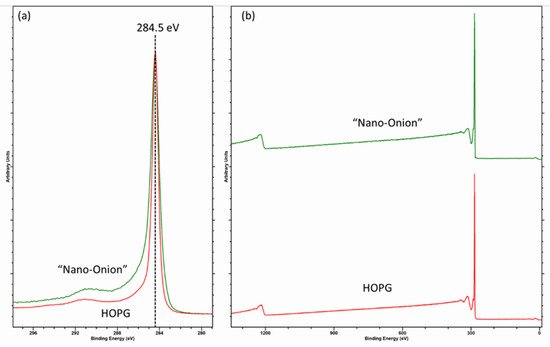
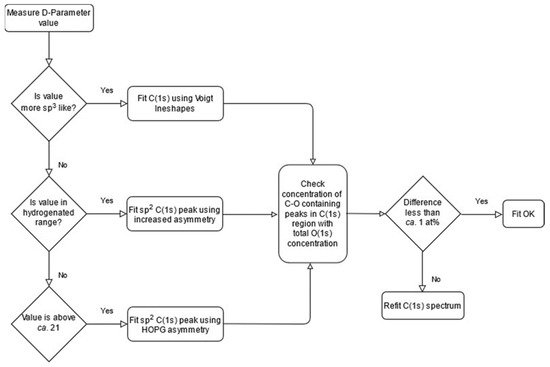
2. The C (1s) Spectrum of Diamond, HOPG and Graphitic Carbons
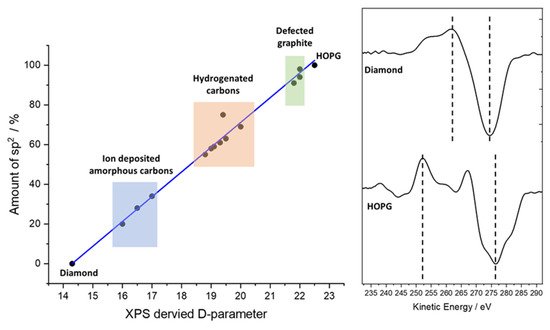
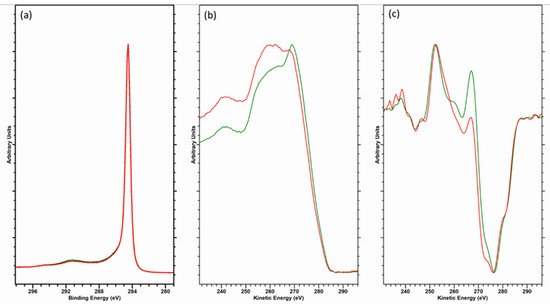
3. The C (KVV) Auger Peak and the D-Parameter
4. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/c7030051
References
- Cazorla-Amorós, D. Grand challenges in carbon-based materials research. Front. Mater. 2014, 1.
- Blanchard, N.P.; Hatton, R.A.; Silva, S.R.P. Tuning the work function of surface oxidised multi-wall carbon nanotubes via cation exchange. Chem. Phys. Lett. 2007, 434, 92–95.
- Butenko, Y.V.; Krishnamurthy, S.; Chakraborty, A.K.; Kuznetsov, V.L.; Dhanak, V.R.; Hunt, M.R.C.; Šiller, L. Photoemission study of onionlike carbons produced by annealing nanodiamonds. Phys. Rev. BCondens. Matter Mater. Phys. 2005, 71.
- Raffa, V.; Ciofani, G.; Nitodas, S.; Karachalios, T.; D’Alessandro, D.; Masini, M.; Cuschieri, A. Can the properties of carbon nanotubes influence their internalization by living cells? Carbon 2008, 46, 1600–1610.
- Jeong, H.K.; Yun, P.L.; Lahaye, R.J.W.E.; Park, M.H.; Kay, H.A.; Ick, J.K.; Yang, C.W.; Chong, Y.P.; Ruoff, R.S.; Young, H.L. Evidence of graphitic AB stacking order of graphite oxides. J. Am. Chem. Soc. 2008, 130, 1362–1366.
- An, K.H.; Yang, C.M.; Seo, K.; Park, K.A.; Lee, Y.H. A diameter-dependent separation of semiconducting from metallic single-wall carbon nanotubes by using nitronium ions. Curr. Appl. Phys. 2006, 6.
- Cheung, T.T.P. X-ray photoemission of carbon: Lineshape analysis and application to studies of coals. J. Appl. Phys. 1982, 53, 6857–6862.
- Ouyang, Y.; Peng, J.C.; Wang, H.; Peng, Z.H. The rehybridization of electronic orbitals in carbon nanotubes. Chinese Phys. B 2008, 17, 3123–3129.
- Chen, X.; Wang, X.; Fang, D. A review on C1s XPS-spectra for some kinds of carbon materials. Full- Nanotub. Carbon Nanostructures 2020, 28, 1048–1058.
- Gengenbach, T.R.; Major, G.H.; Linford, M.R.; Easton, C.D. Practical guides for x-ray photoelectron spectroscopy (XPS): Interpreting the carbon 1s spectrum. J. Vac. Sci. Technol. A 2021, 39, 013204.
- Kaciulis, S. Spectroscopy of carbon: From diamond to nitride films. Surf. Interface Anal. 2012, 44, 1155–1161.
- Kaciulis, S.; Mezzi, A.; Calvani, P.; Trucchi, D.M. Electron spectroscopy of the main allotropes of carbon. Surf. Interface Anal. 2014, 46, 966–969.
- Blume, R.; Rosenthal, D.; Tessonnier, J.P.; Li, H.; Knop-Gericke, A.; Schlögl, R. Characterizing Graphitic Carbon with X-ray Photoelectron Spectroscopy: A Step-by-Step Approach. Chem Cat Chem 2015, 7, 2871–2881.
- Major, G.H.; Avval, T.G.; Moeini, B.; Pinto, G.; Shah, D.; Jain, V.; Carver, V.; Skinner, W.; Gengenbach, T.R.; Easton, C.D.; et al. Assessment of the frequency and nature of erroneous x-ray photoelectron spectroscopy analyses in the scientific literature. J. Vac. Sci. Technol. A 2020, 38, 061204.
- Merlen, A.; Buijnsters, J.G.; Pardanaud, C. A guide to and review of the use of multiwavelength Raman spectroscopy for characterizing defective aromatic carbon solids: From graphene to amorphous carbons. Coatings 2017, 7, 153.
- Wu, J.B.; Lin, M.L.; Cong, X.; Liu, H.N.; Tan, P.H. Raman spectroscopy of graphene-based materials and its applications in related devices. Chem. Soc. Rev. 2018, 47, 1822–1873.
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2004, 362, 2477–2512.
- Lascovich, J.C.; Scaglione, S. Comparison among XAES, PELS and XPS techniques for evaluation of Sp2 percentage in a-C:H. Appl. Surf. Sci. 1994, 78, 17–23.
- Lascovich, J.C.; Giorgi, R.; Scaglione, S. Evaluation of the sp2/sp3 ratio in amorphous carbon structure by XPS and XAES. Appl. Surf. Sci. 1991, 47, 17–21.
- Lascovich, J.C.; Santoni, A. Study of the occupied electronic density of states of carbon samples by using second derivative carbon KVV Auger spectra. Appl. Surf. Sci. 1996, 103, 245–253.
- Lascovich, J.C.; Rosato, V. Analysis of the electronic structure of hydrogenated amorphous carbon via Auger spectroscopy. Appl. Surf. Sci. 1999, 152, 10–18.
- Barlow, A.J.; Scott, O.; Sano, N.; Cumpson, P.J. Multivariate auger feature imaging (MAFI): A new approach towards chemical state identification of novel carbons in XPS imaging. Surf. Interface Anal. 2015, 47, 173–175.
- Barlow, A.J.; Popescu, S.; Artyushkova, K.; Scott, O.; Sano, N.; Hedley, J.; Cumpson, P.J. Chemically specific identification of carbon in XPS imaging using Multivariate Auger Feature Imaging (MAFI). Carbon 2016, 107, 190–197.
- Tanuma, S.; Powell, C.J.; Penn, D.R. Calculations of electron inelastic mean free paths. V. Data for 14 organic compounds over the 50–2000 eV range. Surf. Interface Anal. 1994, 21, 165–176.
- Morgan, D.J. Cluster cleaned HOPG by XPS. Surf. Sci. Spectra 2017, 24, 024003.
- Lesiak, B.; Kövér, L.; Tóth, J.; Zemek, J.; Jiricek, P.; Kromka, A.; Rangam, N. C sp2 /sp3 hybridisations in carbon nanomaterials—XPS and (X)AES study. Appl. Surf. Sci. 2018, 452, 223–231.
- Xpssimplified. Available online: (accessed on 21 June 2021).