Multiple endocrine neoplasia type 1 (MEN1) is a rare autosomal dominant inherited multiple cancer syndrome of neuroendocrine tissues. Tumors are caused by an inherited germinal heterozygote inactivating mutation of the MEN1 tumor suppressor gene, followed by a somatic loss of heterozygosity (LOH) of the MEN1 gene in target neuroendocrine cells, mainly at parathyroids, pancreas islets, and anterior pituitary. Over 1500 different germline and somatic mutations of the MEN1 gene have been identified, but the syndrome is completely missing a direct genotype-phenotype correlation, thus supporting the hypothesis that exogenous and endogenous factors, other than MEN1 specific mutation, are involved in MEN1 tumorigenesis and definition of individual clinical phenotype.
- multiple endocrine neoplasia type 1 (MEN1)
- gene
- loss of heterozygosity (LOH)
- microRNA (miRNAs)
- miR-24
1. Introduction
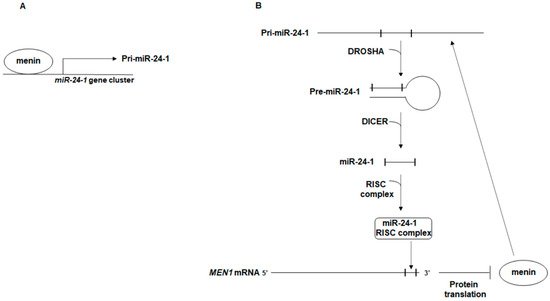
2. The Autoregulatory Network between miR-24, MEN1, and Menin: A Possible Effector of MEN1 Tumorigenesis
| miR-24 Target | Effect | Possible Role in MEN1 Tumorigenesis | Reference |
|---|---|---|---|
| Parathyroid glands | |||
| MEN1 | No effect on MEN1 mRNA expression. Loss of menin protein expression. | Uncontrolled cell proliferation | [27] |
| Endocrine pancreas | |||
| MEN1 | Reduction of both MEN1 mRNA and menin expression. | Uncontrolled cell proliferation | [34] |
| CDKN1B | Reduction of expression of both CDKN1B mRNA and of p27kip1 protein in a mouse insulinoma cell line (MIN6). | Enhanced proliferation of beta-cells and hyperplasia of pancreas islets | [34] |
| CDKN1B | Reduction of expression of CDKN1B mRNA in an immortalized human pancreas beta cell line (Blox5). No data on expression of p27kip1 protein. | Enhanced proliferation of beta-cells and hyperplasia of pancreas islets | [34] |
| CDKN2C | No effect on expression of both CDKN2C mRNA and of p18Ink4c protein in a mouse insulinoma cell line (MIN6). | Non applicable | [34] |
| CDKN2C | Reduction of expression of CDKN2C mRNA in an immortalized human pancreas beta cell line (Blox5). No data on expression of p18Ink4c protein. | Enhanced proliferation of beta-cells and hyperplasia of pancreas islets | [34] |
3. Targeting miR-24: A Potential Therapeutic Tool for MEN1 Tumorigenesis
This entry is adapted from the peer-reviewed paper 10.3390/ijms22147352
References
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L. Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011.
- Brandi, M.L.; Agarwal, S.K.; Perrier, N.D.; Lines, K.E.; Valk, G.D.; Thakker, R.V. Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr. Rev. 2021, 42, 133–170.
- Geslot, A.; Vialon, M.; Caron, P.; Grunenwald, S.; Vezzosi, D. New therapies for patients with multiple endocrine neoplasia type 1. Ann. Endocrinol. 2021, 82, 112–120.
- Al-Salameh, A.; Cadiot, G.; Calender, A.; Goudet, P.; Chanson, P. Clinical aspects of multiple endocrine neoplasia type 1. Nat. Rev. Endocrinol. 2021, 17, 207–224.
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–407.
- Pannett, A.A.; Thakker, R.V. Somatic mutations in MEN type 1 tumors, consistent with the Knudson “two-hit” hypothesis. J. Clin. Endocrinol. Metab. 2001, 86, 4371–4374.
- Valdes, N.; Alvarez, V.; Diaz-Cadorniga, F.; Aller, J.; Villazon, F.; Garcia, I.; Herrero, A.; Coto, E. Multiple endocrine neoplasia type 1 (MEN1): LOH studies in an affected family and in sporadic cases. Anticancer Res. 1998, 18, 2685–2689.
- Dreijerink, K.M.A.; Timmers, H.T.M.; Brown, M. Twenty years of menin: Emerging opportunities for restoration of transcriptional regulation in MEN1. Endocr. Relat. Cancer 2017, 24, T135–T145.
- Feng, Z.; Ma, J.; Hua, X. Epigenetic regulation by the menin pathway. Endocr. Relat. Cancer 2017, 24, T147–T159.
- Marini, F.; Giusti, F.; Tonelli, F.; Brandi, M.L. Pancreatic Neuroendocrine Neoplasms in Multiple Endocrine Neoplasia Type 1. Int. J. Mol. Sci. 2021, 22, 4041.
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670.
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402.
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233.
- Shukla, G.C.; Singh, J.; Barik, S. MicroRNAs: Processing, Maturation, Target Recognition and Regulatory Functions. Mol. Cell Pharmacol. 2011, 3, 83–92.
- Tüfekci, K.U.; Meuwissen, R.L.J.; Genç, S. The role of microRNAs in biological processes. Methods Mol. Biol. 2014, 1107, 15–31.
- Vidigal, J.A.; Ventura, A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2015, 25, 137–147.
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, 15004.
- Lin, Y.-C.; Tso-Hsiao Chen, T.-H.; Huang, Y.-M.; Wei, P.-L.; Lin, J.-C. Involvement of microRNA in Solid Cancer: Role and Regulatory Mechanisms. Biomedicines 2021, 9, 343.
- Tan, W.; Liu, B.; Qu, S.; Liang, G.; Luo, W.; Gong, C. MicroRNAs and cancer: Key paradigms in molecular therapy. Oncol. Lett. 2018, 15, 2735–2742.
- Bouyssou, J.M.; Manier, S.; Huynh, D.; Issa, S.; Roccaro, A.M.; Ghobrial, I.M. Regulation of microRNAs in cancer metastasis. Biochim. Biophys. Acta 2014, 1845, 255–265.
- Jafri, M.A.; Al-Qahtani, M.H.; Shay, J.W. Role of miRNAs in human cancer metastasis: Implications for therapeutic intervention. Semin. Cancer Biol. 2017, 44, 117–131.
- Nagy, Z.; Szabó, P.M.; Grolmusz, V.K.; Perge, P.; Igaz, I.; Patócs, A.; Igaz, P. MEN1 and microRNAs: The link between sporadic pituitary, parathyroid and adrenocortical tumors? Med. Hypotheses 2017, 99, 40–44.
- Grolmusz, V.K.; Borka, K.; Kövesdi, A.; Németh, K.; Balogh, K.; Dékány, C.; Kiss, A.; Szentpéteri, A.; Sármán, B.; Somogyi, A.; et al. MEN1 mutations and potentially MEN1-targeting miRNAs are responsible for menin deficiency in sporadic and MEN1 syndrome-associated primary hyperparathyroidism. Virchows Arch. 2017, 471, 401–411.
- Luzi, E.; Pandolfini, L.; Ciuffi, S.; Marini, F.; Cremisi, F.; Nesi, G.; Brandi, M.L. MicroRNAs regulatory networks governing the epigenetic landscape of MEN1 gastro-entero-pancreatic neuroendocrine tumor: A case report. Clin. Transl. Med. 2021, 11, e351.
- Donati, S.; Ciuffi, S.; Marini, F.; Palmini, G.; Miglietta, F.; Aurilia, C.; Brandi, M.L. Multiple Endocrine Neoplasia Type 1: The Potential Role of microRNAs in the Management of the Syndrome. Int. J. Mol. Sci. 2020, 21, 7592.
- Lal, A.; Navarro, F.; Maher, C.A.; Maliszewski, L.E.; Yan, N.; O’Day, E.; Chowdhury, D.; Dykxhoorn, D.M.; Tsai, P.; Hofmann, O.; et al. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3’UTR microRNA recognition elements. Mol. Cell 2009, 35, 610–625.
- Luzi, E.; Marini, F.; Giusti, F.; Galli, G.; Cavalli, L.; Brandi, M.L. The negative feedback-loop between the oncomir Mir-24-1 and menin modulates the Men1 tumorigenesis by mimicking the “Knudson’s second hit”. PLoS ONE 2012, 7, e39767.
- Evers, B.M.; Ishizuka, J.; Townsend, C.M., Jr.; Thompson, J.C. The human carcinoid cell line, BON. A model system for the study of carcinoid tumors. Ann. N. Y. Acad. Sci. 1994, 733, 393–406.
- Luzi, E.; Marini, F.; Ciuffi, S.; Galli, G.; Brandi, M.L. An autoregulatory network between menin and pri-miR-24-1 is required for the processing of its specific modulator miR-24-1 in BON1 cells. Mol. Biosyst. 2016, 12, 1922–1928.
- Gao, J.; Zhu, M.; Liu, R.-F.; Zhang, J.-S.; Xu, M. Cardiac Hypertrophy is Positively Regulated by MicroRNA-24 in Rats. Chin. Med. J. 2018, 131, 1333–1341.
- Hall, C.; Ehrlich, L.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Lairmore, T.C.; Alpini, G.; Glaser, S. Inhibition of microRNA-24 increases liver fibrosis by enhanced menin expression in Mdr2(−/−) mice. J. Surg. Res. 2017, 217, 160–169.
- Qiaoqiao, C.; Li, H.; Liu, X.; Yan, Z.; Zhao, M.; Xu, Z.; Wang, Z.; Shi, K. MiR-24-3p regulates cell proliferation and milk protein synthesis of mammary epithelial cells through menin in dairy cows. J. Cell Physiol. 2019, 234, 1522–1533.
- Alrezk, R.; Hannah-Shmouni, F.; Stratakis, C.A. MEN4 and CDKN1B mutations: The latest of the MEN syndromes. Endocr. Relat. Cancer. 2017, 24, T195–T208.
- Vijayaraghavan, J.; Maggi, E.C.; Crabtree, J.S. miR-24 regulates menin in the endocrine pancreas. Am. J. Physiol Endocrinol. Metab. 2014, 307, E84–E92.
- Ehrlich, L.; Hall, C.; Venter, J.; Dostal, D.; Bernuzzi, F.; Invernizzi, P.; Meng, F.; Trzeciakowski, J.P.; Zhou, T.; Standeford, H.; et al. miR-24 Inhibition Increases Menin Expression and Decreases Cholangiocarcinoma Proliferation. Am. J. Pathol. 2017, 187, 570–580.
- Pan, Y.; Wang, H.; Ma, D.; Ji, Z.; Luo, L.; Cao, F.; Huang, F.; Liu, Y.; Dong, Y.; Chen, Y. miR-24 may be a negative regulator of menin in lung cancer. Oncol. Rep. 2018, 39, 2342–2350.
- Montero, C.; Sanjuán, P.; del Mar Fernández, M.; Vidal, I.; Verea, H.; Cordido, F. Bronchial carcinoid and type 1 multiple endocrine neoplasia syndrome. A case report. Arch. Bronconeumol. 2010, 46, 559–561. (In Spanish)
- Gang, D.; Hongwei, H.; Hedai, L.; Ming, Z.; Qian, H.; Zhijun, L. The tumor suppressor protein menin inhibits NF-κB-mediated transactivation through recruitment of Sirt1 in hepatocellular carcinoma. Mol. Biol. Rep. 2013, 40, 2461–2466.
- Gallo, A.; Agnese, S.; Esposito, I.; Galgani, M.; Avvedimento, V.E. Menin stimulates homology-directed DNA repair. FEBS Lett. 2010, 584, 4531–4536.
- Jin, S.; Mao, H.; Schnepp, R.W.; Sykes, S.M.; Silva, A.C.; D’Andrea, A.D.; Hua, X. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003, 63, 4204–4210.
- Gambari, R.; Brognara, E.; Spandidos, D.A.; Fabbri, E. Targeting oncomiRNAs and mimicking tumor suppressor miRNAs: Νew trends in the development of miRNA therapeutic strategies in oncology (Review). Int. J. Oncol. 2016, 49, 5–32.