p38 mitogen-activated protein kinases (MAPKs) are key signaling molecules in cellular responses to external stresses and regulation of pro-inflammatory cytokines. Some studies have suggested that p38 MAPK in the region of the nucleus accumbens is involved in abnormal behavioral responses induced by drugs of abuse. In this entry, we recapitulate the role of the p38 MAPK in the rewarding effects of drugs of abuse. We also summarize the implication of p38 MAPK in stress, anxiety, and depression. We opine that p38 MAPK activation is more closely associated to stress-induced aversive responses rather than drug effects per se, in particular cocaine. p38 MAPK is only involved in cocaine reward, predominantly when promoted by stress. Downstream substrates of p38 that may contribute to the p38 MAPK associated-behavioral responses are proposed. Finally, we suggest p38 MAPK inhibitors as possible therapeutic interventions against stress-related disorders by potentially increasing resilience against stress and addiction relapse induced by adverse experiences.
- p38 MAPK
- cocaine
- conditioned place preference
- reward
- stress
- anxiety
- depression
- nucleus accumbens
- social interaction
- k opioid receptors
1. Introduction
The mitogen-activated protein kinase (MAPK) is a serine/threonine kinase that is activated through phosphorylation by a MAPK kinase (MKK), which is a “dual-specific” kinase that phosphorylates at both serine/threonine and tyrosine residues within a threonine/any amino acid/tyrosine (Thr X Tyr) motif, in which the middle amino acid is different for each MAPK [1]. Enzymes in the p38 MAPK module are subject to dual phosphorylation at the Thr-Gly-Tyr motif situated within the kinase activation loop and are primarily activated by various environmental stresses, including heat, osmotic and oxidative stresses, as well as inflammatory cytokines [2] (Figure 1).
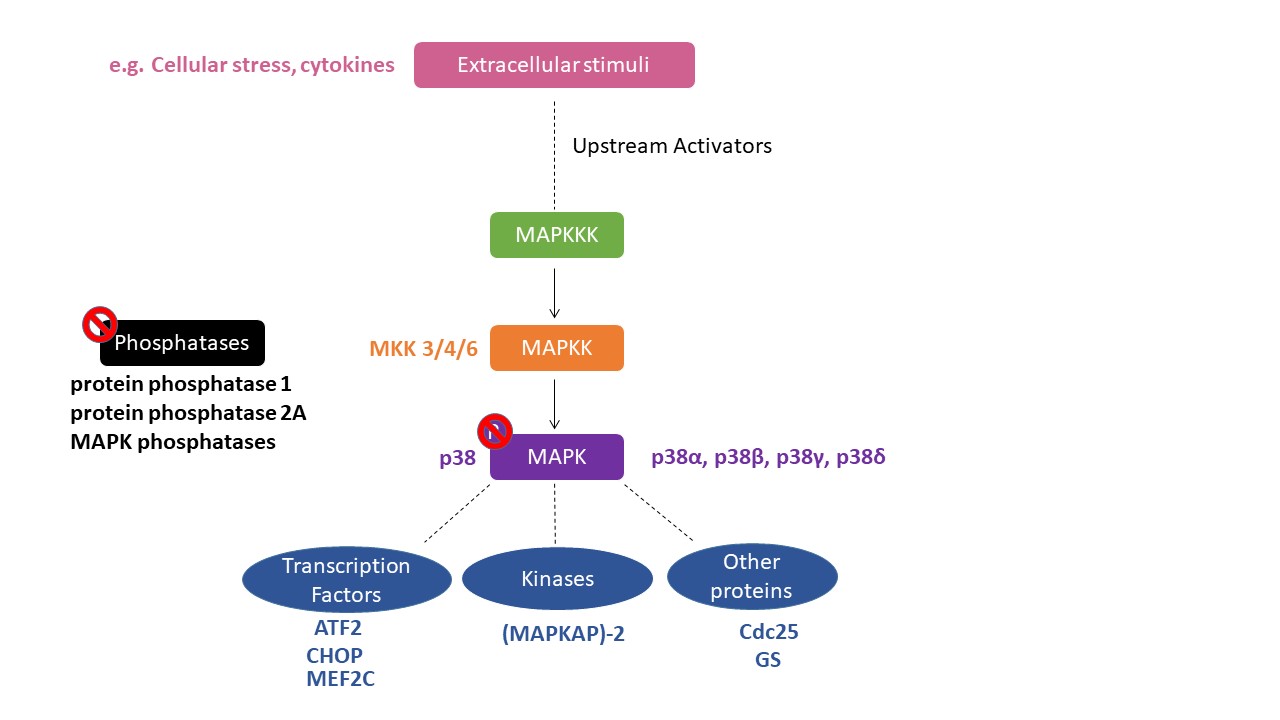
2. Role of p38 MAPK in the Rewarding Effects of Drugs of Abuse
Some studies have suggested that p38 MAPK in the region of the nucleus accumbens (NAc) is involved in abnormal behavioral responses induced by drugs of abuse. The rewarding effects of drugs of abuse can be assessed by conditioned place preference (CPP) in which animals can associate drugs' effects to a specific context, and "prefer" to spend more time in the drug-associated compartment when the drug is pleasurable (Figure 2).
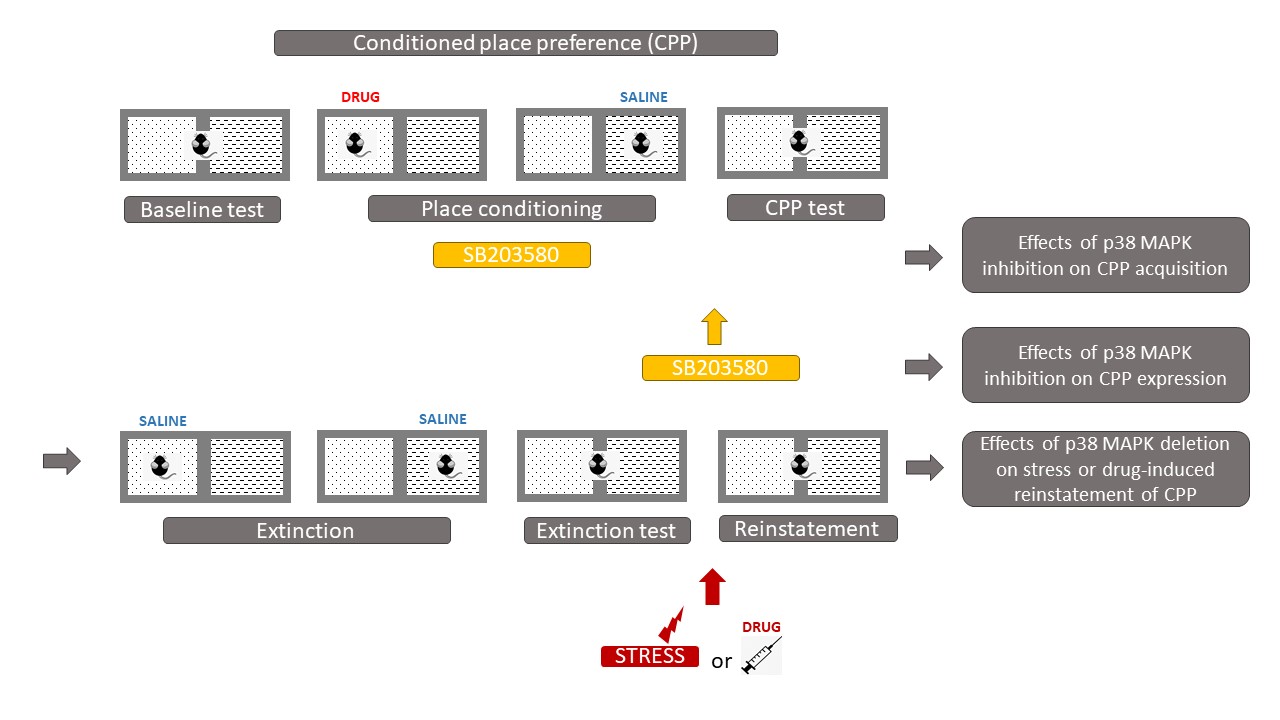
Gerdjikov et al. showed that NAc injections of the p38 kinase inhibitor SB203580 dose-dependently impaired acquisition of amphetamine CPP [3]. Moreover, several studies showed that repeated morphine treatment increased the phosphorylation of p38 in the NAc [4] [5]. Consistently, the microinjection of SB203580 into the NAc prior to the administration of morphine or intraperitoneal (i.p.) injections of SB203580 prevented the acquisition of CPP and inhibited the activation of p38 [4] [5] [6] . Following the acquisition of morphine CPP, a SB203580 injection failed to block the expression of morphine CPP [5] [7]. At the contrary to morphine, cocaine CPP does not induce p38 activation in the NAc [8]. Moreover, p38 activation in the NAc shell sub-division is decreased by social interaction reward, when social interaction is paired with one compartment of the CPP [8]. Studies that performed intracerebroventricular (icv) injections of SB203580 before cocaine training found no effect on cocaine CPP [9] [10]. Consistently, the deletion of p38α in the serotonergic neurons of the dorsal raphe nucleus (DRN) [11] or the conditional knock-out of p38α MAPK in the dopaminergic neurons of the ventral tegmental area (VTA) [12] does not affect cocaine CPP, suggesting that the deletion of p38α does not alter the associative learning required for place preference or the rewarding properties of cocaine. Interestingly, serotonergic p38α MAPK deletion blocked the reinstatement of cocaine preference induced by stress, but not by cocaine priming, thereby showing that serotonergic p38α MAPK deletion selectively alters only the stress-induced modulation of cocaine-seeking behaviors [11]. Therefore, p38 is considered to be more closely related to stress modulation than to cocaine treatment. Indeed, rats receiving corticotropin-releasing factor (CRF) icv injections before cocaine conditioning showed an increase in cocaine CPP, whereas those receiving icv injections of the non-selective antagonist alpha-helical CRF (α CRF) showed a decrease in cocaine CPP [10]. Remarkably, when social interaction was made available in the alternative compartment, CRF-induced increase of cocaine preference was reversed completely to the level of rats receiving cocaine paired with α CRF. This reversal of cocaine preference was also paralleled by a reversal in altered behavioral sequencing of grooming, considered as a marker of stress [13] and by a CRF-induced increase of p38 MAPK expression in the NAc shell [10]. These results show that social interaction has anti-stress effects and that the modulation of the CRF system has a direct impact on p38 MAPK expression in the NAc shell, as p38 MAPK expression was increased after icv CRF injections prior to each cocaine conditioning and was decreased after icv injections of α CRF prior to each cocaine conditioning [10].
3. Role of p38 MAPK in Stress, Anxiety, and Depression
Sustained stressful experience can lead to maladaptive responses, including clinical depression, anxiety, and an increased risk for drug addiction [14] [15]. After stress exposure, p38 MAPK is generally activated in different brain regions. Repeated forced swim stress activated p38 MAPK in the cortex, the hippocampus, and the NAc [9], which decreased significantly following a pretreatment with SB203580 before bouts of forced swimming [9]. pp38 levels were also increased in the prefrontal cortex (PFC) after cold exposure [16], in the hippocampus after enhanced single prolonged stress [17] and in the DRN after social defeat stress [11]. Additionally, a significant positive correlation was found between early life stress and the percentage of monocytes staining positive for pp38 [18]. Neuro-inflammation, in response to bacterial endotoxin lipopolysaccharide (LPS)-induced depressive-like behaviors, has been reported to be accompanied by increased levels of pp38 in the habenula [19]. Both the p38 inhibitor SB203580 and the anti-depressant fluoxetine normalized the changes in p38 phosphorylation and reversed the depressive-like behaviors [19]. Interestingly, the depletion of neuronal p38α in mice resulted specifically in increased anxiety-related behaviors without affecting learning and memory processes or motor coordination and muscle function [20]. p38 activation appears to be an important mediator of KOR-induced aversive stress effects through G-protein-coupled receptor kinase 3 (GRK3)/β-arrestin, a KOR-associated protein, dependent mechanisms [9] [21]. Inhibition of p38 MAPK was found to block stress-induced behavioral responses, including aversive responses to the KOR agonist U50, 488 [9] [22]. Importantly, cell-specific deletion of p38α MAPK in serotonergic neurons blocked stress-induced aversion [11]. These effects seem to be regulated by KOR as KOR knockout (KO) mice did not develop conditioned place aversion (CPA) to U50,488; however, re-expression of KOR in the serotonergic neurons of the DRN or in the dopaminergic neurons of the VTA of KOR KO mice activated p38 and restored place aversion [12] [23].
4. Possible substrates encoding the behavior responses to stress downstream to p38 MAPK
One possible mechanism encoding the behavior responses to stress is a change in gene expression downstream to p38 [9]. One candidate is zif268, whose induction is p38-dependent [9]. Another possible substrate of pp38 that may contribute to the behavioral responses is the serotonin transporter (SERT). Stress-induced p38α MAPK caused translocation of SERT to the plasma membrane in the brain, thereby increasing the rate of transmitter uptake at serotonergic nerve terminals and inducing a hypo-serotonergic state that underlies depression-like and drug-seeking behaviors [11]. A third possible candidate could involve the modulation of proteins related to synaptic plasticity, such as AMPA receptors. p38 MAPK signaling has been shown to be an important mediator of AMPA receptor surface trafficking during synaptic plasticity in which activation of p38 MAPK may lead to synaptic removal of surface AMPA receptors [24] [25]. One additional target of p38 relevant to depression could involve the glucocorticoid receptor (GR). p38 signaling pathways have been shown to be implicated in the inhibition of GR function. Indeed, activation of p38 MAPK has been demonstrated to disrupt transactivation of the GR [26], leading potentially to glucocorticoid resistance or decreased responsiveness to glucocorticoids, a primary feature of major depression [27]. Figure 3 summarizes the possible substrates that may contribute to the behavioral responses associated to p38 MAPK.
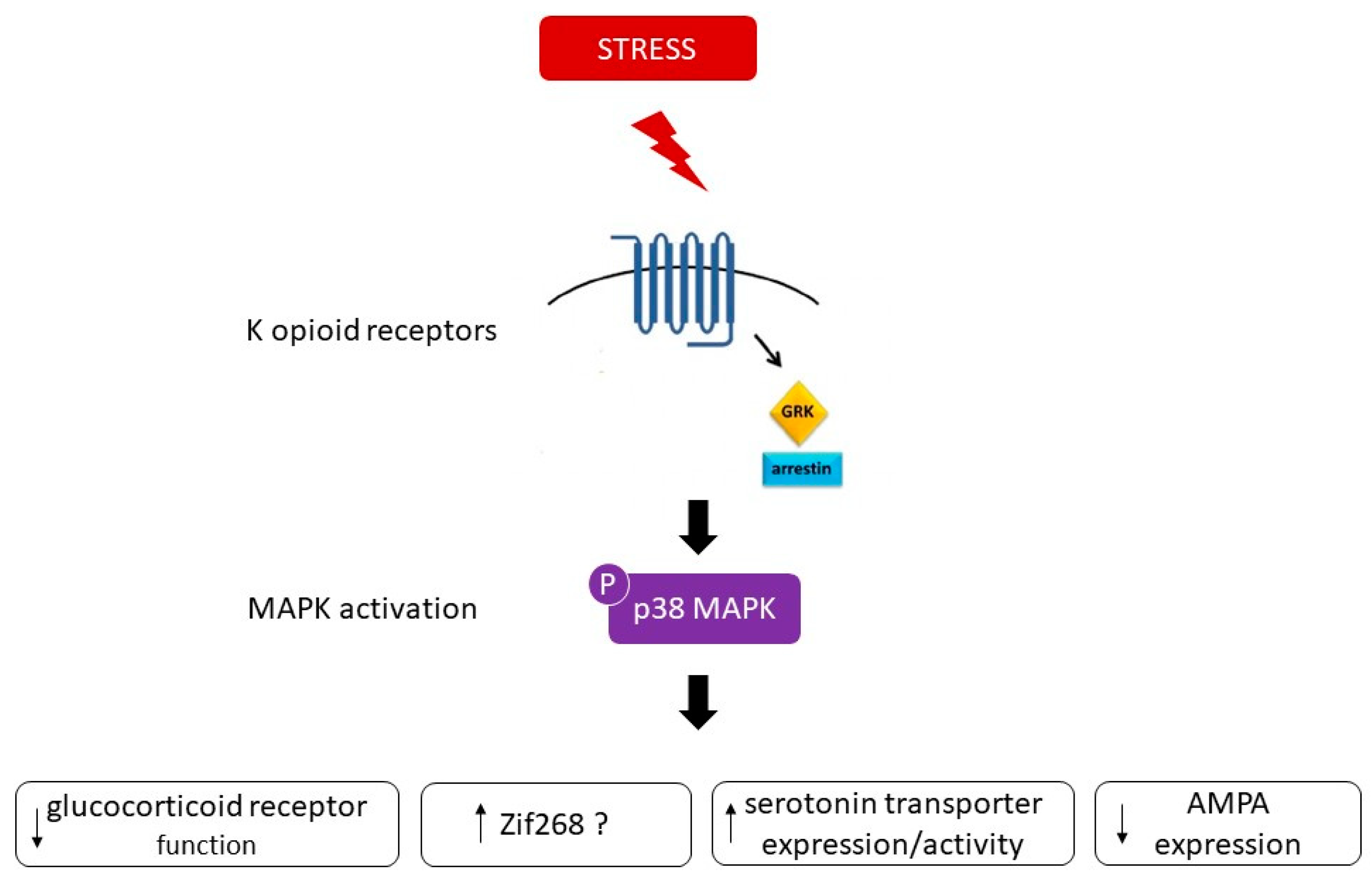
Figure 3. Arrestin-dependent signaling events result in p38 MAPK activation and subsequent dysphoric behavioral responses. Possible substrates encoding the behavior responses to stress downstream to p38 comprises changes in gene expression such as zif268, in serotonin transporter expression/activity, in glucocorticoid receptor function and modulation of proteins related to synaptic plasticity, such as AMPA receptors.
5. Conclusion
It appears that p38 MAPK activation is more closely associated to stress-induced aversive responses rather than drug effects per se. Mostly, studies show that p38 MAPK activation is only involved in cocaine reward, predominantly when promoted by stress. However, it remains open to discussion how p38 MAPK is implicated in CPP morphine acquisition. It could be that morphine might activate KOR as well as µ opioid receptors (MOR) or that MOR opioids could to some extent induce activation of p38. In conclusion, understanding the molecular and cellular mechanisms that control stress-induced behaviors could explain the neurobiological mechanisms involved in depression and addiction-like behaviors and provides insight to potential therapeutic targets. Indeed, p38 MAPK inhibitors have been studied extensively in both preclinical experiments and clinical trials for inflammatory diseases. Here, we opine that p38 MAPK inhibitors are of growing interest as possible therapeutic interventions against stress-related disorders by potentially increasing resilience against stress and addiction relapse induced by adverse experiences.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21144833
References
- K. J. Cowan; Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. The Journal of Experimental Biology 2003, 206, 1107-1115, 10.1242/jeb.00220.
- Liguo New; Jiahuai Han; The p38 MAP Kinase Pathway and Its Biological Function. Trends in Cardiovascular Medicine 1998, 8, 220-228, 10.1016/s1050-1738(98)00012-7.
- Todor V. Gerdjikov; Gregory M. Ross; Richard J. Beninger; Place Preference Induced by Nucleus Accumbens Amphetamine Is Impaired by Antagonists of ERK or p38 MAP Kinases in Rats.. Behavioral Neuroscience 2004, 118, 740-750, 10.1037/0735-7044.118.4.740.
- Xueqin Zhang; Yue Cui; Jin Jing; Wenjun Xin; Xian-Guo Liu; Yu Cui; Involvement of p38/NF-κB signaling pathway in the nucleus accumbens in the rewarding effects of morphine in rats. Behavioural Brain Research 2011, 218, 184-189, 10.1016/j.bbr.2010.11.049.
- Xue-Qin Zhang; Yu Cui; Yue Cui; Yu Chen; Xiao-Dong Na; Feng-Ying Chen; Xu-Hong Wei; Yong-Yong Li; Xian-Guo Liu; Wen-Jun Xin; et al. Activation of p38 signaling in the microglia in the nucleus accumbens contributes to the acquisition and maintenance of morphine-induced conditioned place preference. Brain, Behavior, and Immunity 2012, 26, 318-325, 10.1016/j.bbi.2011.09.017.
- Sa-Ik Hong; Thi-Lien Nguyen; Shi-Xun Ma; Hyoung-Chun Kim; Seok-Yong Lee; Choon-Gon Jang; TRPV1 modulates morphine-induced conditioned place preference via p38 MAPK in the nucleus accumbens. Behavioural Brain Research 2017, 334, 26-33, 10.1016/j.bbr.2017.07.017.
- Padmanabhan Mannangatti; Kamalakkannan Narasimhanaidu; Mohamad Imad Damaj; Sammanda Ramamoorthy; Lankupalle Damodara Jayanthi; A Role for p38 Mitogen-activated Protein Kinase-mediated Threonine 30-dependent Norepinephrine Transporter Regulation in Cocaine Sensitization and Conditioned Place Preference. Journal of Biological Chemistry 2015, 290, 10814-10827, 10.1074/jbc.m114.612192.
- Ahmad Salti; Kai Kummer; Chinmaya Sadangi; Georg DeChant; Alois Saria; Rana El Rawas; Social interaction reward decreases p38 activation in the nucleus accumbens shell of rats.. Neuropharmacology 2015, 99, 510-6, 10.1016/j.neuropharm.2015.08.029.
- Michael R. Bruchas; Benjamin B. Land; Megumi Aita; Mei Xu; Sabiha K. Barot; Shuang Li; C Chavkin; Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria.. The Journal of Neuroscience 2007, 27, 11614-23, 10.1523/JNEUROSCI.3769-07.2007.
- Cristina Lemos; Ahmad Salti; Inês Amaral; Veronica Fontebasso; Nicolas Singewald; Georg DeChant; Alex Hofer; Rana El Rawas; Social interaction reward in rats has anti-stress effects.. Addiction Biology 2020, ., e12878, 10.1111/adb.12878.
- Michael R. Bruchas; Abigail G. Schindler; Haripriya Shankar; Daniel I. Messinger; Mayumi Miyatake; Benjamin B. Land; Julia C. Lemos; Catherine E. Hagan; John F. Neumaier; Albert Quintana; et al. Selective p38α MAPK Deletion in Serotonergic Neurons Produces Stress Resilience in Models of Depression and Addiction. Neuron 2011, 71, 498-511, 10.1016/j.neuron.2011.06.011.
- Jonathan M. Ehrich; Daniel I. Messinger; Cerise R. Knakal; Jamie R. Kuhar; Selena S. Schattauer; Michael R. Bruchas; Larry Zweifel; Brigitte L. Kieffer; Paul E. M. Phillips; Charles Chavkin; et al. Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons.. The Journal of Neuroscience 2015, 35, 12917-31, 10.1523/JNEUROSCI.2444-15.2015.
- Allan V. Kalueff; Pentti Tuohimaa; The grooming analysis algorithm discriminates between different levels of anxiety in rats: potential utility for neurobehavioural stress research. Journal of Neuroscience Methods 2005, 143, 169-177, 10.1016/j.jneumeth.2004.10.001.
- Rajita Sinha; Chronic stress, drug use, and vulnerability to addiction.. Annals of the New York Academy of Sciences 2008, 1141, 105-30, 10.1196/annals.1441.030.
- Nuno Sousa; The dynamics of the stress neuromatrix. Molecular Psychiatry 2016, 21, 302-312, 10.1038/mp.2015.196.
- Gang Zheng; Yaoming Chen; Xueping Zhang; Tongjian Cai; Mingchao Liu; Fang Zhao; Wenjing Luo; Jingyuan Chen; Acute cold exposure and rewarming enhanced spatial memory and activated the MAPK cascades in the rat brain. Brain Research 2008, 1239, 171-180, 10.1016/j.brainres.2008.08.057.
- Z Peng; R Zhang; F Xue; H Nie; Y Chen; D Wu; Y Wang; H Wang; Q Tan; Gastrodin ameliorates anxiety-like behaviors and inhibits IL-1beta level and p38 MAPK phosphorylation of hippocampus in the rat model of posttraumatic stress disorder.. Physiological Research 2013, 62, 537-545, 10.33549/physiolres.932507.
- M M Sanchez; O Alagbe; J C Felger; J Zhang; A E Graff; A P Grand; D Maestripieri; A H Miller; Activated p38 MAPK is associated with decreased CSF 5-HIAA and increased maternal rejection during infancy in rhesus monkeys. Molecular Psychiatry 2007, 12, 895-897, 10.1038/sj.mp.4002025.
- Ya-Wei Zhao; Yu-Qin Pan; Ming-Ming Tang; Wen-Juan Lin; Blocking p38 Signaling Reduces the Activation of Pro-inflammatory Cytokines and the Phosphorylation of p38 in the Habenula and Reverses Depressive-Like Behaviors Induced by Neuroinflammation. Frontiers in Pharmacology 2018, 9, 511, 10.3389/fphar.2018.00511.
- Kristie Stefanoska; Josefine Bertz; Alexander M. Volkerling; Julia Van Der Hoven; Lars M. Ittner; Arne Ittner; Neuronal MAP kinase p38α inhibits c-Jun N-terminal kinase to modulate anxiety-related behaviour. Scientific Reports 2018, 8, 14296, 10.1038/s41598-018-32592-y.
- Benjamin B. Land; Michael R. Bruchas; Julia C. Lemos; Mei Xu; Erica J. Melief; C Chavkin; The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system.. The Journal of Neuroscience 2008, 28, 407-14, 10.1523/JNEUROSCI.4458-07.2008.
- G.-Y. Zan; Q. Wang; Y.-J. Wang; J.-C. Chen; X. Wu; C.-H. Yang; J.-R. Chai; M. Li; Y. Liu; X.-W. Hu; et al. p38 mitogen-activated protein kinase activation in amygdala mediates κ opioid receptor agonist U50,488H-induced conditioned place aversion. Neuroscience 2016, 320, 122-128, 10.1016/j.neuroscience.2016.01.052.
- Benjamin B. Land; Michael R. Bruchas; Selena Schattauer; William J. Giardino; Megumi Aita; Daniel Messinger; Thomas S. Hnasko; Richard D. Palmiter; Charles Chavkin; Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proceedings of the National Academy of Sciences 2009, 106, 19168-19173, 10.1073/pnas.0910705106.
- Chiung-Chun Huang; Jia-Lin You; Mei-Ying Wu; Kuei-Sen Hsu; Rap1-induced p38 Mitogen-activated Protein Kinase Activation Facilitates AMPA Receptor Trafficking via the GDI·Rab5 Complex. Journal of Biological Chemistry 2004, 279, 12286-12292, 10.1074/jbc.m312868200.
- Ying-Ching Liang; Chiung-Chun Huang; Kuei-Sen Hsu; A role of p38 mitogen-activated protein kinase in adenosine A1 receptor-mediated synaptic depotentiation in area CA1 of the rat hippocampus. Molecular Brain 2008, 1, 13-13, 10.1186/1756-6606-1-13.
- X Wang; H Wu; A H Miller; Interleukin 1α (IL-1α) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Molecular Psychiatry 2003, 9, 65-75, 10.1038/sj.mp.4001339.
- Thaddeus W.W. Pace; Fang Hu; Andrew H. Miller; Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain, Behavior, and Immunity 2007, 21, 9-19, 10.1016/j.bbi.2006.08.009.