1. Insulin Signaling in Metabolism and Circadian Rhythm Regulation
Insulin/IGF–1 pathway activity plays a major role in the control of aging [
74,
75,
76,
77,
78] and in the beneficial effects of CR [
79,
80]. Insulin is a key regulator of glucose uptake and utilization in insulin–responsive tissues. Following food intake, increased blood glucose levels trigger pancreatic β–cells to secrete insulin. Free circulating insulin activates insulin receptors on the surface of target cells eliciting a signaling cascade initiated by the activation of insulin receptor substrates (IRS 1–4) followed by phosphorylation of phosphoinositide 3–kinase (PI3K), which manages metabolic response including PDK1 and Akt stimulation by phosphorylation. Akt signaling prompts glucose transporter 4 (GLUT4) to translocate to the cell membrane where it initiates cellular glucose uptake. Akt also stimulates the production of glycogen and inhibits gluconeogenesis. Moreover, Akt activates mTOR, which facilitates anabolic processes, while mTORC2 feeds back to regulate Akt [
81]. Insulin signaling affects multiple downstream pathways including mitogen–activated protein kinase (MAPK), which controls growth, sterol regulatory element–binding protein 1 (SREBP–1), which stimulates the synthesis of lipid and cholesterol as well as the family of Forkhead (FOXO) transcription factors regulating metabolism and autophagy [
82,
83]. Inhibition of IGF–1/PI3K/Akt signaling contributes to the anti–cancer and DNA–repair activity of CR [
84,
85,
86].
The direct interconnection between CR and circadian rhythms has been evinced by the fact that
Igf–1 expression is regulated by both CR and the circadian clock [
64]. Interestingly,
Igf–1 expression is rhythmic and shows sexual dimorphism [
64]. Furthermore, a genome–wide RNAi screen for genes that regulate cellular clock functions in human cells identified the insulin signaling pathway as the most overrepresented pathway [
87]. Accordingly, the impact of CR on plasma IGF–1 and insulin level is compromised in mice deficient for
Bmal1 [
71]. Further, the postprandial release of insulin resets peripheral clocks by regulating the expression of core circadian genes. Insulin rapidly increases the expression of
Per2 in insulin–sensitive tissues like the liver, muscle, or adipose tissue, but not the lung or brain [
88]. Insulin secreted upon refeeding–after–fasting stimulates
Per2 and reduces
Rev–erbα expression in hepatocytes [
27]. The capacity of insulin to initiate entrainment of the liver clock was demonstrated by the administration of insulin in cultured rat hepatocytes which acutely induced expression of
Per1,
Per2, and
Dec1 [
88]. Accordingly, inhibition of pathways downstream of insulin signaling, such as MAPK and PI3K, blocks the induction of the
Per1 and
Per2 clock genes [
88].
Next to insulin, glucose is a circadian clock regulator. Glucose levels control AMPK activity, which phosphorylates and controls the stability of the CRY proteins [
89]. In rats, glucose infusion during the light phase strongly induces expression of
Per2 in the SCN and reduces
Per2 expression in the liver [
90]. Accordingly, a reduced level of glucose availability delays the light–induced phase shift [
91]. Glucose also reduces the expression of
Per1 and
Per2 in fibroblasts in vitro [
92].
Reciprocally, the circadian clock in the pancreas regulates insulin and glucagon production and secretion and their signaling in SCN via the autonomic nervous system [
93,
94,
95,
96]. Glucagon secretion is also regulated by Rev–Erb
α [
94]. In vitro islet β–cells exhibit robust rhythms of both
Bmal1 and
Per1 [
95,
97], while disruption of circadian clock functions in pancreas–specific
Bmal1 KO mice leads to glucose intolerance, defective insulin production and secretion [
95]. BMAL1 and CLOCK contribute to the regulation of the recovery from the hypoglycemic response to insulin [
98]. Mice deficient in BMAL1 and CLOCK exhibit dysregulated glucose homeostasis, impaired glucose tolerance, and reduced insulin sensitivity [
93]. On the contrary, KO of the negative arm of the circadian machinery, that is invalidation of
Crys,
Pers, or Rev–erbα leads to increased insulin levels [
99,
100,
101]. In the
Per2 KO mouse, insulin secretion is more effectively stimulated by glucose and its analogs compared to WT animals. At the same time, the circadian rhythm of hepatic insulin–degrading enzyme (Ide) is disrupted leading to decreased insulin clearance. Consequently,
Per2 KO animals suffer from hyperglycemia [
100].
Control of glucose homeostasis requires both the central and peripheral clocks, and disruption of synchronization between them affects glucose metabolism negatively [
102]. CLOCK drives the transcriptional stimulation of glycogen synthase 2 (
Gys–2) and therefore modulates the circadian rhythms of hepatic glycogen synthesis [
103]. BMAL1 and CLOCK stimulate gluconeogenesis that consequently is reduced in the KO models of these proteins [
98]. Similarly, RORα directly induces phosphoenolpyruvate–carboxykinase (
Pepck) expression [
104] and thus, RORα deficiency, as well as treatment with RORα antagonists, inhibits PEPCK expression and glucose production [
105]. Accordingly, glucose–6–phosphatase (G6Pase) and PEPCK are suppressed in HepG2 cells overexpressing
Rev–erbα, encoding the physiological repressor of RORα. Accordingly, silencing
Rev–erbα significantly increases the expression of G6Pase and PEPCK [
104,
106,
107]. Alike, during fasting, rhythmically expressed CRY proteins in the liver reduce gluconeogenesis through phosphorylation of cAMP response element–binding protein (CREB) [
108], and phosphoenolpyruvate carboxykinase 1 (PCK1) by direct interaction of CRY with the
Pck1 promoter [
109]. Furthermore, during feeding and acute fasting, PER2 dampers gluconeogenesis and enhances glycogen storage by decreasing the activity of glycogen phosphorylase (GP) [
110].
2. mTOR Signaling and the Circadian Rhythm
The mTOR pathway is a key effector pathway of CR and it is known for monitoring the availability of nutrients and regulating longevity. TOR is one of the Ser/Thr protein kinases from the family of phosphatidylinositol 3 (PI3) kinase–related kinases [
111,
112] and it functions as a key component of two complexes, mTORC1 and mTORC2 [
111]. mTORC1 is sensitive to cellular energy levels, nutrient status, mitogenic signals, and oxygen levels and it is inhibited by rapamycin. mTORC1 signaling leads to the regulation of mRNA translation and autophagy. mTORC2 is not rapamycin sensitive and operates as a regulator of the cellular actin cytoskeleton [
113,
114]. The mTOR pathway integrates intracellular and extracellular physiological stimuli. In this pathway, the protein complex of tuberous sclerosis proteins 1 and 2 (TSC1 and TSC2) mediates upstream signals from growth factors, such as insulin and IGF–1 to mTORC1. Activation of mTOR mediates the phosphorylation of several executor proteins involved in mRNA translation and ribosome biogenesis, such as ribosomal S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E–binding protein (4E–BP) [
115,
116,
117]. mTORC1 downstream signaling controls autophagy and metabolism, including the glycolytic turnover and anabolic processes associated with the fed state including lipogenesis [
118,
119,
120,
121], cholesterol synthesis via activation of SREBP–1/2 [
118,
122,
123], and protein synthesis [
124,
125].
mTOR activity in vivo is induced by the abundance of nutrients and gradually decreases during fasting. However, food–independent rhythmicity in activity and expression of the mTORC1 complex members has been observed in the SCN and liver, cardiac and skeletal muscles, adipocytes, and retinal photoreceptors but not in the intestine or lung [
126,
127,
128,
129,
130,
131,
132,
133,
134]. In the mouse brain, mTORC1 activities exhibit daily alterations in the arcuate nucleus, hippocampus, and the frontal cortex [
130,
135,
136], all regions that manage circadian rhythms, feeding, learning, memory, and emotions. The oscillations of mTOR activity are delimited by internal and external regulators [
137]. In the brain of
Drosophila, TOR rhythms are found particularly in the ventral lateral neurons [
138,
139]. Neuronal TORC1 and AKT signaling have been shown to drive behavior [
138]. Also, in the SCN, mTORC1 signaling is activated by light and controls behavior in a circadian manner [
126,
127,
140]. Brief light exposure of mice during the night, but not during the day, triggers instant phosphorylation of the mTOR translation effectors S6K1, S6 ribosomal protein (S6), and translational repressor 4E–BP1 [
126]. A KO of 4E–BP1 in mice leads to a higher amplitude of molecular rhythms in the SCN, increased capacity for re–entrainment to a shifted light/dark cycle, and higher resistance to the disruption of rhythm by constant light [
140]. In vivo, infusion of the inhibitor of mTOR1 rapamycin leads to an attenuation of the phase–delaying effect of early–night light. Equally, disruption of mTOR during the late–night augments the phase–advancing effect of light. At both the early– and late–night time points, abrogation of mTOR signaling leads to a significant attenuation of light–induced PER protein expression [
141]. Conversely, constitutive activation of mTOR in
Tsc2–deficient fibroblasts alters the dynamics of clock gene rhythmicity and raises levels of principal clock proteins, including CRY1, BMAL1, and CLOCK [
142]. Heterozygous
mTor KO mice present a lengthened circadian period of locomotor activity rhythms both in constant darkness and constant light [
142]. mTOR inhibition lengthens the period and dampens the amplitude of circadian clock proteins, whereas mTOR activation shortens the period and augments amplitude in hepatocytes, adipocytes, and human U2OS cells [
142,
143]. In
Drosophila, TOR modulates the circadian period in opposite direction compared to mice. Overexpressing S6K in the ventral lateral neurons, the central pacemaker cells, extends the circadian period [
138]. Consistently, KO of
Tor in Per expressing cells shortens the period of locomotor rhythms in
Drosophila [
139].
Importantly, the intracellular concentration of Mg
2+ ions act as a cell–autonomous timekeeping component that controls key clock properties from unicellular algae to human. Furthermore, Mg
2+ ions oscillations regulate circadian control of translation by mTOR [
143].
In the liver, fasting/feeding cycles regulate the energy sensor AMPK and mTORC1 [
28,
129,
130] independently of CRY1 and CRY2 but partly relying on BMAL1 [
137]. S6K1 rhythmically phosphorylates BMAL1 at an evolutionarily conserved site to activate it for modulation of mRNA translation and protein synthesis. Therefore, protein synthesis rates show circadian oscillations that are dependent on BMAL1 [
131], which deficiency increases mTORC1 activity both in vivo and in vitro [
130]. Increased mTOR signaling correlates with faster aging while treatment with rapamycin extends the lifespan of prematurely–aging
Bmal1 KO mice by 50% [
130]. Therefore, increased BMAL1 activity likely contributes to lower mTORC1 activity in CR. In summary, the regulation of mTOR downstream and upstream of the circadian rhythm connects daily oscillations with sensing of the nutrient status (
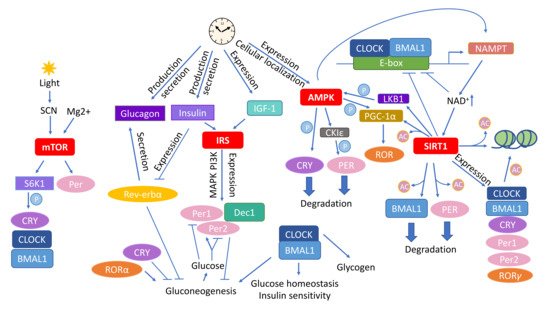
Figure 2).
Figure 2. Circadian rhythm affects the main molecular pathways mediating the outcome of caloric restriction (CR). Various types of interactions connect the CR–associated pathways mTOR, insulin signaling, AMPK, and sirtuins with circadian rhythm. Abbreviations: AMPK—adenosine monophosphate (AMP) activated protein kinase; CKIε—casein kinase ε; CLOCK—circadian locomotor output cycles kaput; Dec1—differentially expressed In chondrocytes 1; IGF–1—insulin–like growth factor 1; IRS—insulin receptor substrates; LKB1—liver kinase B1; mTOR—mammalian target of rapamycin; NAD—nicotinamide adenine dinucleotide; NAMPT—nicotinamide phosphoribosyltransferase; SIRT1—Sirtuin 1; SCN—suprachiasmatic nucleus, S6K1—ribosomal protein S6 kinase 1. The circled Ac indicates acetylation and circled P phosphorylation.
3. AMPK as an Energy Sensor and Executor of Circadian Metabolic Activities
AMPK serves as a key energy sensor of the AMP:ATP and ADP:ATP ratios. AMPK, activated at low levels of ATP, suppresses energy–consuming anabolic pathways and promotes catabolism resulting in the recruitment of stored energy to reestablish the ATP levels [
144]. AMPK impacts metabolism via several downstream effectors including CREB–regulated transcriptional coactivator–2 (CRTC2) [
145], TBC1D1/AS160 [
146,
147], peroxisome proliferator–activated receptor γ coactivator 1 α (PGC–1α) [
148], and histone deacetylase 5 (HDAC5) [
149]. Functionally, AMPK inhibits fatty acid (FA), TG and cholesterol synthesis, while stimulating FA uptake and β–oxidation [
150,
151,
152,
153,
154]. Furthermore, AMPK reduces protein synthesis by inhibiting mTOR [
155]. AMPK also affects the metabolism of glucose by stimulating glycolysis [
156] and inhibiting glycogen synthesis [
157] and gluconeogenesis [
158,
159,
160]. It also drives nutrient–induced insulin secretion from islet β–cells [
161] and glucose uptake by GLUT4 [
162]. Besides metabolism, AMPK plays a role in inflammation, cell growth, autophagy, and apoptosis [
163].
The circadian clock is paired with cellular metabolic fluctuations via nutrient–sensing pathways. While the expression of six of the transcripts encoding subunits of mammalian AMPK does not vary during the day, that of the AMPKβ2 subunit is eight times higher in the middle of the day than it is at night. AMPKβ2 determines the cellular localization of the heterotrimeric AMPK complex [
164]; therefore, the presence of the AMPKα1 subunit in the nucleus increases at the time of day when AMPKβ2 expression peaks [
89]. The rhythmic hepatic activity and nuclear localization of AMPK correlate inversely with CRY1 levels, which is likely due to the fact that stimulation of AMPK leads to phosphorylation and degradation of CRY [
89]. Further, activation of AMPK by 5–aminoimidazole–4–carboxamide ribonucleoside (AICAR) or metformin in mouse livers causes a phase shift of the clock, and animals in which the AMPK pathway is genetically disrupted show alterations in peripheral clocks [
89,
165]. Moreover, acute AICAR stimulation alters the expression of clock genes in WT mice but not in mice lacking the AMPKγ3 regulatory subunit [
166]. Genetic disruption of either AMPKα1 or AMPKα2 subunit dampens the rhythm of body temperature, the free–running activity, changes the circadian pattern of core clock gene expression in mice in an isoform– and tissue–specific manners [
165]. In mouse muscles, AMPK regulates the expression patterns of the circadian genes
Cry2,
Rev–erbα, and
Bhlhb2 (basic helix–loop–helix domain containing class B 2) [
166]. Moreover, AMPK is capable of phosphorylating casein kinase ε (CKIε) and thereby increases its enzymatic activity, indirectly leading to a destabilization of PER2 [
167]. PGC–1α, which co–activates the RORs and consequently stimulates the expression of
Bmal1 and
Rev–erbα, is phosphorylated by AMPK [
148,
168,
169]. PGC–1α is required for cell–autonomous clock function [
169] and PGC–1α KO mice show an abnormal diurnal rhythm of physical activity, body temperature, and metabolic rate, due to disrupted expression of clock genes and genes involved in energy metabolism. Besides the direct impact on ATP levels, AMPK affects the energy status by promoting feeding through signaling in the hypothalamus as well as by adjusting circadian metabolism [
170,
171]. AMPK may thereby mediate the influence of fasting/feeding cycles on the circadian clock (
Figure 2).
4. SIRT Energy Sensors in the Context of CR and Daily Rhythmicity
SIRTs serve as energy sensors by detecting the ratio of reduced to oxidized nicotinamide adenine dinucleotide NAD
+:NADH and react as transcriptional effectors mostly through their HDAC activity. SIRTs are class III HDACs that manage processes connected with nucleic acid biology including DNA repair, homologous recombination, and histone deacetylation, as well as transcriptional gene silencing [
172,
173]. There are seven subtypes of SIRTs (SIRT1–7) in mice and humans which differ in their cellular localization and function. SIRT1–SIRT3, SIRT5, SIRT6, and SIRT7 act as deacetylases, while SIRT4 and SIRT6 have ADP–ribosylation activity. Besides histones, SIRTs modify also several transcriptional regulators including the nuclear factor kappa–light–chain enhancer of activated B cells (NF–κB), p53, FOXO, PGC–1α, as well as enzymes, including acetyl coenzyme A (CoA) synthetase 2 (AceCS2), long–chain acyl–coenzyme A dehydrogenase (LCAD), 3–hydroxy–3–methylglutaryl–CoA synthase 2 (HMGCS2), superoxide dismutase 2 (SOD–2), and structural proteins, such as α–tubulin [
174,
175,
176,
177,
178]. Therefore, SIRTs impacts multiple processes and pathways including circadian clocks, cell cycle, mitochondrial biogenesis, and energy homeostasis, consequently influencing aging, apoptosis, inflammation, and stress resistance [
179,
180]. SIRT1 is mostly associated with metabolism. In
S. cerevisiae an extra copy of the Sir2 gene, a yeast homolog of mammalian Sirt1, increases lifespan [
181,
182], whereas the deletion of Sir2 shortens it [
181]. In yeast and
Drosophila, lack of the Sirt1 homolog offsets CR–triggered life extension [
183,
184,
185]. A yeast analog of Sirt1 takes part in DNA repair and regulates aging–related gene expression [
186].
PGC–1α, one of the main metabolic effectors of SIRT1, is activated by SIRT1–mediated deacetylation [
187,
188]. Activated PGC–1α enhances hepatic gluconeogenesis [
187], mitochondrial activity in muscle and BAT leading to increased exercise capacity and thermogenesis; consequently, PGC–1α promotes protection against obesity and metabolic dysfunction [
189]. SIRTs interact also with factors involved in response to CR including the FOXO family of transcription factors [
190,
191,
192], which affects gluconeogenesis and glucose release from hepatocytes [
193], cell differentiation, metabolism as well as longevity regulation [
194,
195,
196]. Further, AMPK enhances SIRT1 activity by increasing cellular NAD
+ levels [
197,
198] and activation of SIRT1 may cause AMPK phosphorylation via deacetylation–dependent activation of the AMPK–activating kinase liver kinase B1 (LKB1) [
199,
200].
SIRT1 controls the circadian expression of the core clock genes
Bmal1,
Rorγ,
Per2, and
Cry1. Also, SIRT1 is recruited by CLOCK:BMAL1 chromatin at circadian promoters [
201]. Further, the
levels of NAD
+, NADP
+, NADH, and NADPH affect the binding capacity of CLOCK–BMAL1 heterodimers to E–box elements [
202]. Similarly, resveratrol, a polyphenolic SIRT activator, regulates the expression of clock genes
Per1,
Per2, and
Bmal1 in Rat–1 fibroblast cells [
203]. It also modifies the rhythmic expression of clock genes (
Clock,
Bmal1, and
Per2) and lipid metabolism–related genes controlled by the clock (
Pparα,
Srebp–1c,
Acc1, and
Fas) in HFD–fed mice [
204] and reverses the change induced by high–fat feeding in the expression of Rev–Erbα in adipose tissue of rats [
205]. SIRT1 also promotes the deacetylation and subsequent degradation of PER2 in a circadian manner [
201].
The rhythmic acetylation of BMAL1 and acetyl–histone H3 Lys9/Lys14 at circadian promoters correlates with SIRT1 HDAC activity that is regulated in a circadian manner. Therefore, genetic or pharmacological inhibition of SIRT1 activity causes disturbances in the acetylation of H3 and BMAL1 and the circadian rhythm [
206]. Moreover, the circadian transcription factor CLOCK has histone acetyltransferase (HAT) activity, and SIRT1 HDAC activity counteracts the HAT activity of CLOCK [
201,
206]. Another circadian protein, REV–ERBα regulates pancreatic glucagon secretion via the AMPK/nicotinamide phosphoribosyltransferase (NAMPT)/SIRT1 pathway [
94]. Further, the expression of NAMPT, a rate–limiting enzyme involved in NAD
+ production through the salvage pathway, is regulated by the BMAL/CLOCK heterodimer. Consequently, NAD
+ levels exhibit rhythmic daily oscillations [
207,
208]. Moreover, by being recruited to the
Nampt promoter, SIRT1 contributes to the circadian synthesis of its own coenzyme [
208]. Inhibition of NAMPT promotes fluctuations of
Per2 by releasing CLOCK:BMAL1 from suppression by SIRT1. In turn, CLOCK binds to the
Nampt promoter and stimulates its activity, thereby contributing to a feedback loop comprising NAMPT/NAD
+ and SIRT1/CLOCK:BMAL1 [
207].
SIRT3 also interacts with the circadian rhythm. It sets the pace in the acetylation and activity of oxidative enzymes and consequently respiration in isolated mitochondria.
Bmal1 KO mice have significantly decreased SIRT3 activity, which affects mitochondrial oxidative function, and supplementation with nicotinamide mononucleotide (NMN), a NAD
+ precursor, restores SIRT3 function and enhances oxygen consumption in these animals [
209]. Importantly, the rhythm of cyclic global protein acetylation dampens with aging in mice [
70]. CR regulates SIRT1 activity and therefore it modulates the circadian acetylation of AceCS1, a pathway controlling rhythmic nucleocytoplasmic acetyl–CoA production [
210,
211]. Consequently, CR rescues the hepatic protein acetylation rhythm over the day/night cycle in mice. Accordingly, the circadian transcriptome of CR–mediated effects on circadian reprogramming and SIRT1–specific transcriptome overlaps [
70] indicating a pivotal role of SIRT1 in connecting CR and the circadian rhythm (
Figure 2).