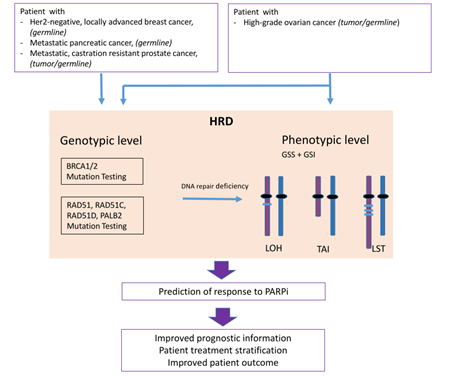
Homologous Recombination Deficiency (HRD) is associated with sensitivity towards PARP inhibitors (PARPi) and its determination is used as a biomarker for therapy decision making.
1. Introduction
One major underlying mechanism is the emergence of genomic instability caused by genetic mutations arising from exogenously or endogenously caused DNA damage or failures in DNA damage repair. Healthy cells maintain genomic integrity by a variety of repair mechanisms, each addressing unique forms of DNA damage. The base excision repair (BER) pathway is activated by damaged DNA bases (DNA single strand-lesions). In response to double-strand breaks, two different repair pathways are available; the exact mechanism of homologous recombination repair (HRR) and the error-prone non-homologous end joining (NHEJ) [2].
Defects in the DNA repair system are an underlying cause of genomic instability due to the accumulation of genetic changes [3]. Homologous recombination repair deficiency (HRD) resulting in DNA double-strand breaks is considered to be the most lethal of all DNA repair defects since cancer cells switch to the error-prone NHEJ pathway fostering genomic instability and cell death. PARP inhibition (PARPi) disables single-strand break repair and leads to further accumulation of double-strand breaks, consequently requiring homologous recombination repair. Albeit improved outcome results led to the approval of several inhibtors for different tumor entities, it remains challenging to define those patients who might benefit from PARPi therapy.
2. DNA Repair Mechanisms
Mammalian cells obey different cellular responses to DNA damage, including DNA damage repair, cell cycle arrest and apoptosis. Regarding the responses of DNA repair, different mechanisms can take place. The simplest way of DNA repair is proofreading or direct reversal repair, occurring during replication. The methyl group is transferred to a catalytically active cysteine and afterwards MGMT is targeted to the proteasome for degradation [4].Nevertheless, it requires more sophisticated repair mechanisms to effectively correct complex damage on already replicated DNA caused by several endogenous or exogenous factors.
Depending on the factor that causes DNA damage, different types of mismatches/errors occur on single-stranded DNA. For example, radiation modifies single bases or DNA adducts alter the conformation of the DNA. Mismatches created during the replication process due to slippage of DNA strands are corrected by the so-called mismatch repair. Mismatched bases or reading frame shifts are recognised by specific protein dimers, consisting of MSH2 and MSH3 or MSH6.
DNA damage of single bases caused by oxidation, deamination or alkylation are commonly repaired by base excision repair (BER). In a first step, the damaged base is recognised and removed by specialized DNA glycosylases. At this point, the enzyme called Poly (ADP-ribose) polymerase (PARP) binds to the gapped DNA strand and engages proteins required for strand protection and repair. Following this, the DNA strand is sealed by ligases in complex with X-ray repair cross-completing protein 1 (XRCC1) in the short-patch BER.
Two major repair mechanisms have evolved to deal with double-strand lesions, homologous recombination repair (HRR) and non-homologous end joining (NHEJ). The nature of DSBs caused by internal factors, such as replication block, and external influences, such as ionizing radiation, is obviously different. On one hand, lesions associated with the replication process can be directly repaired by using the sister chromatid, which is located in close proximity, as a template for homologous recombination (HR). In order to deal with such types of lesions on DNA in condensed chromatin structures, vertebrates frequently use NHEJ to simply re-ligate the DSB end strands.
Homologous recombination repair (HRR) is an accurate repair mechanism to cope with DSBs. In a first step, the lesion is recognised by the MRE11-RAD50-NBS1 (MRN) complex, which activates ATM (Ataxia telangiectasia mutated) kinase. Upon DNA 5´-end resection, the replication protein (RPA) coats the single-strand DNA regions and activates ATR (Ataxia telangiectasia and Rad3-related) kinase. In the following, RPA is replaced by RAD51 with the help of further repair-associated proteins, such as CHEK2, BRCA1, BRCA2 and PALB2, which are loaded with RAD51. Subsequently, the defective DNA strand attaches to its sister chromatid, which is used as a template for DNA resynthesis. This mechanism of DSB repair is restricted to late S- and G2 phases of the cell cycle [9,10]
The second pathway that evolved to cope with DSBs belongs to the simplest mechanisms: non-homologous end joining (NHEJ). The core component of this pathway is the Ku70–Ku80 complex, which is able to bind ends of double-strand broken DNA and recruit DNA-PKcs to initiate NHEJ. In a final step, DNA ligase IV (LIG4) is simultaneously recruited with XRCC4 and an XRCC4-like factor (XLF) to ligate the processed DNA and restore genome integrity. Thereby, chromosomal integrity is prone to get lost, giving rise to chromosomal rearrangements [2,11].
The impairment of HRR activity is called homologous recombination repair deficiency (HRD) and is caused by different factors. Additionally, expression of the HRR associated proteins can be diminished by promoter methylation, for example [12]. In order to deal with this impairment, cells tend to activate the alternative NHEJ pathway for DSB repair. As already mentioned, this pathway is prone to ending in chromosomal rearrangements.
The most common pathogenic alterations of HRR associated genes occur within the genes BRCA1and BRCA2[14]. Nevertheless, other genes of the pathway and their associated proteins can be affected as well. This phenotype of homologous recombination repair deficiency independent of BRCA1/2 mutations is referred to as BRCAness [10].
2. PARP Inhibition and Homologous Recombination Repair Deficiency
2.1. The Concept of Synthetic Lethality
Utilizing poly (ADP-ribose) polymerase (PARP) inhibitors in BRCA deficient cells has shown great promise in clinical studies for patients with
BRCA1/2 mutated tumors. Meanwhile, several PARP inhibitors have been approved for the treatment of ovarian, breast, prostate and pancreatic cancers in different clinical settings. With the recent approval of the combination of olaparib and bevacizumab as a maintenance treatment for high-grade epithelial ovarian cancer based on the data of the PAOLA-1 trial, HRD positivity was added as a new biomarker for patient stratification [
15]. For metastatic castration-resistant prostate cancer (mCRPC), olaparib received FDA approval in patients with either mutations in
BRCA1, BRCA2, ATM or one out of 12 other HR-related genes. This indication was based on the results gathered from the PROfound trial [
16].
PARP1 and PARP2 add long chains of poly (ADP-ribose) to several proteins and PARP itself, a process which is called PARylation. PARP inhibitors prevent the modification of PARP proteins, thereby impeding dissociation of PARP from the DNA single-strand break. Furthermore, attachment of additional repair proteins is inhibited, leading to accumulation of single-strand breaks. In replicating cells these single-strand breaks are converted into double-strand breaks [
17]. Besides the inhibition of single-strand repair, PARP inhibitors bind PARP1 and PARP2 enzymes to damaged DNA. This so-called PARP trapping holds a strong cytotoxic effect. PARP-DNA complexes result in a stalled replication fork and further DNA double-strand breaks accumulate. In healthy cells these double-strand breaks are eliminated by the mechanism of homologous recombination repair (HRR) [
18].
Cells with HRD caused by alterations in the genes encoding HR proteins are forced to use the error-prone pathway of non-homologous end joining (NHEJ). This leads to the accumulation of genomic damage, thus PARP inhibition is particularly effective in the absence of an intact HR pathway [
2,
19]. DNA double-strand breaks associated with PARP inhibition and replication lead to chromosomal rearrangement, genomic instability and apoptotic cell death. The concept of combining two conditions to force cell death is known as synthetic lethality and its use in tumors with defective DNA repair or altered checkpoint controls was described by Ashworth in 2008 for the first time [
20]. Initially, only mutations in
BRCA1 and
BRCA2 were introduced as predictive biomarkers for successful PARP inhibition. Interestingly, in a significant proportion of tumors holding HRD, variants within these two genes could not be detected [
15]. Therefore, analysis of genomic instability as a read-out for alterations in the HR pathway was additionally proven as a suitable biomarker for a response to PARP inhibition.
2.2. Biomarkers for PARPi: Genes Involved in the Homologous Recombination Repair Pathway
The most common alterations currently known to confer sensitivity to PARP inhibition are loss of function mutations in
BRCA1 and
BRCA2 [
21].
BRCA1 can also be inhibited by gene promoter methylation but, regarding responses to PARP inhibitor therapy, retrospective and preclinical data showed conflicting results. In ovarian cancer patients,
BRCA1 methylation was not associated with long-term responses [
22], whereas, in patient-derived xenografts, a homozygous
BRCA1 methylation status led to PARP inhibitor sensitivity [
23].
Additionally, preclinical data suggested that patients whose tumors show HRD caused by other mutations in the HR pathway, may benefit from therapy with PARP inhibitors [
18]. Several different types of proteins are involved in protecting genome integrity and present possible biomarkers for PARP inhibition. Kinases, such as ATM or ATR, are responsible for damage recognition; a second group are signal mediators, such as CHEK2 and BRCA1; finally, repair is initiated by effector proteins, such as BRCA2 and RAD51. Other proteins, such as PALB2 and BRIP1, function as facilitators of the HR pathway [
15].
The prevalence of mutations in HR genes, apart from
BRCA1 and
BRCA2, in all solid tumors was largely unknown and differed between published data. Therefore, Heeke et al. (2018) carried out a comprehensive molecular profiling in a large cohort of more than 17,000 solid tumor specimens [
14]. Their study revealed pathogenic mutations in HR pathway genes in 17.4% of tumors with endometrial cancer being most commonly mutated (34.4%), followed by biliary tract (28.9%), bladder (23.9%), hepatocellular (20.9%) and ovarian cancer (20.0%). The most frequently mutated HR genes included
ARID1A (7.2%),
BRCA2 (3.0%),
BRCA1 (2.8%),
ATM (1.3%),
ATRX (1.3%) and
CHEK2 (1.3%). Several of these mutations correlate with clinical responses to PARP inhibitor treatment. Recently, it was shown that patients with
PALB2 mutated metastatic breast cancer benefit from PARP inhibition in contrast to patients with exclusive
ATM or
CHEK2 mutations [
24]. Seventeen percent to twenty-five percent of pancreatic cancers harbor mutations in genes related to DNA repair [
25] and recent clinical trials suggested the benefit of PARP inhibitor treatment for patients with platinum-sensitive pancreatic cancer genomic alterations in DNA repair genes beyond
BRCA [
26].
Mutational analysis of
BRCA1, BRCA2 and additional HR-related genes can be performed on either blood for germline testing or tissue samples for both germline and somatic testing. The decision, whether germline or somatic mutation status, has to be determined depending on the specific approval of a PARP inhibitor for a certain tumor entity [
27]. Tissue testing is routinely carried out on formalin-fixed paraffin-embedded tissue (FFPE), thus the extracted DNA may be strongly degraded and insufficient for the detection of large gene deletions. Moreover, FFPE tissue testing is hampered by the occurrence of fixation artifacts, which may lead to false-positive results. On the other hand, blood testing only detects germline mutations and is not suitable for simultaneous analysis of the HRD phenotype.
The above described alterations can be detected by parallel sequencing with targeted gene panels. Different pre-assembled panels are commercially available from different providers, but a custom design is also possible. Whereas the detection of these mutations seems to be technically manageable, interpretation of mutations in the HR-related genes in a clinical context remains challenging. For
BRCA variants, the internationally agreed classification scheme of the International Agency for Research on Cancer (IARC) [
28], the regulations of the ENIGMA consortium (Evidence-based Network for the Interpretation of Germline Mutant Alleles) and databases, such as
BRCA Exchange [
29], aid in variant classification. For non-
BRCA variants, the same classification scheme can be used [
30], but databases are currently being assembled and still include a high number of variants of unknown significance.
Interpreting the consequences of alterations in HR-related genes in different tumor entities remains challenging and clinical data show that mutations in specific genes can be of predictive value for only certain entities [
31]. The underlying DNA repair mechanisms are complex, thus the role of a single protein as a predictive biomarker depends largely on its specific function within the repair process. Additionally, the low frequency of the individual mutations in different tumor entities complicates the assessment of their relevance. Considering the current state of knowledge testing for alterations in HR-related genes should always include a bunch of those genes and single gene approaches should be avoided.
The assessment of the predictive value of mutations in HR-related genes beyond
BRCA1 and
BRCA2 depends on the evaluation of further prospectively collected data. Whereas the FDA has already approved the use of PARP inhibitor therapy for mCRPC with mutations in HR-related genes based on the data of the PROfound trial [
16], the respective ESMO working group stated in their recommendations that the evidence for clinical validity of non-
BRCA HRR genes is currently too low [
21].
2.3. Biomarker for PARPi: Genomic Instability
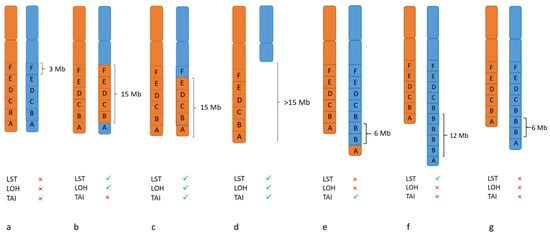
LOH- TAI- LST
BRCA1 and
BRCA2 mutations are commonly used to identify patients with HRD. In a recent clinical trial in ovarian cancer it was shown that nearly 20% of the study population was HRD positive without having a
BRCA mutation [
15]. As these patients also benefit from PARP inhibition, assays identifying HRD without knowing the underlying mechanisms should be applied. HRD causes patterns of mutations and deletions/insertions (mutational signatures) as well as copy number variations (CNV) and structural rearrangements, effects which can be analyzed by different molecular methods. The larger genome alterations are called “genomic scars“ [
32] and are currently the basis of available clinical assays for HRD, which can only be performed on tumor tissue. These genomic scars comprise loss of heterozygosity (LOH), telomeric allelic imbalance (TAI) and large scale state transitions (LST). LOH occurs by either deletion of one allele (copy loss LOH) or by deletion and simultaneous duplication of the remaining allele (copy neutral LOH), resulting in the loss of one of the two alleles at a heterozygous locus. TAI, telomeric allelic imbalance, is defined as the number of regions with allelic imbalance extending to the subtelomere but not crossing the centromere. (
Figure 1) [
33,
34,
35]. HRD-related genomic alterations have to be distinguished from other genomic alterations found in cancer genomes. Thus, measures for these three markers in the context of HR-deficient tumors have been defined using single nucleotide polymorphism (SNP) arrays in the respective cohorts (
Figure 1). Abkevich and colleagues (Br. J. Cancer, 2012) showed that the number of subchromosomal segments with LOH of a size exceeding 15 Mb but shorter than the length of a whole chromosome is associated with a functional inactivation of BRCA1, BRCA2 or RAD51C [
33]. Telomeric allelic imbalances extend from the double-strand breakpoint to the subtelomeric region of a chromosome without including the centromere. High levels of such aberrations are shown in tumors with BRCA1 and BRCA2 deficiency as well as tumors sensitive to platinum chemotherapy [
34]. LST is defined as a third signature by Popova et al., (Cancer Res. 2012) as the number of break points occurring between adjacent regions of at least 10 Mb. This marker was established in breast tumors and cell lines with BRCA deficiency [
35].
Figure 1. Examples for different alterations which lead to genomic scars. (a) Normal chromosomal pattern with maternal and paternal alleles (A–F); each box represents 3 Mb. (b–g) Alterations like rearrangements, loss or gain of chromosomal material that can result in positive scores for LOH (loss of heterozygosity), LST (large scale state transitions) and TAI (telomeric allelic imbalance).
LOH, TAI and LST were found to be independently associated with homologous repair deficiency, but the combination of all three scores allows for the most robust prediction [
36]. A composite HRD score was developed in three TNBC (triple negative breast cancer) clinical trial cohorts using the unweighted sum of the three single scores. Furthermore, a threshold for HRD positivity was selected based on the likelihood of response to platinum-based chemotherapy [
37].
A major limitation of the genomic scar assays is the potential lack of representing the current HRD status when analyzing archival tumor tissue. Tumor cells previously defined to be HR-deficient might have restored their proficiency by resistance mechanisms, such as reversion mutations in HR-related genes. RAD51 focus formation or replication fork assays could be used as functional HRD assays. Nevertheless, there are still some challenges to be solved prior to implementing such assays into routine diagnostics, such as the need for fresh tissue and standardization of positivity thresholds [
38].
Commercially available tests most often combine
BRCA mutation testing with either a composite HRD score or the assessment of LOH, as described within the manuscript (
Table 1). To date, only the Myriad myChoice assay and the Foundation Focus CDx BRCA LOH assay were used for patient stratification in clinical studies [
39]. Thus, alternative assays must be validated with clinical samples previously analyzed with one of the above-mentioned assays.
Beyond BRCA1 and BRCA2 mutations, alterations in other HR-related genes, as well as inactivation by promoter methylation and large genomic defects, so-called genomic scars, are used as biomarkers to stratify patients for PARP inhibitor therapies. Currently, it seems that this large variety of biomarkers and tests used for the assessment remain limited in their ability to effectively identify all patients who might benefit from PARP inhibitor treatment (Figure 3).
This entry is adapted from the peer-reviewed paper 10.3390/jpm11070612