In the last two decades, the therapeutic landscape of several tumors have changed profoundly with the introduction of drugs against proteins encoded by oncogenes. Oncogenes play an essential role in human cancer and when their encoded proteins are inhibited by specific drugs, the tumoral process can be reverted or stopped. An example of this is the case of the chronic myeloid leukemia, in which all the pathological features can be attributed by a single oncogene. Most patients with this disease now have a normal life expectancy thanks to a rationality designed inhibitor. However, the drug only blocks the protein, the oncogene continues unaffected and treatment discontinuation is only an option for a small subset of patients. With the advent of genome-editing nucleases and, especially, the CRISPR/Cas9 system, the possibilities to destroy oncogenes now is feasible. A novel therapeutic tool has been developed with unimaginable limits in cancer treatment. Recent studies support that CRISPR/Cas9 system could be a definitive therapeutic option in chronic myeloid leukemia. This work reviews the biology of chronic myeloid leukemia, the emergence of the CRISPR system, and its ability as a specific tool for this disease.
- CML
- CRISPR/Cas9
- BCR/ABL1
- genome editing
1. Clinical Features of Chronic Myeloid Leukemia
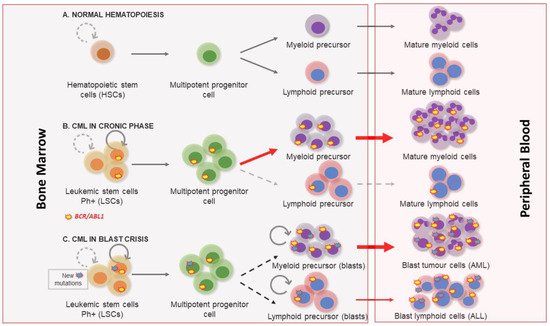
2. Molecular Biology of Chronic Myeloid Leukemia
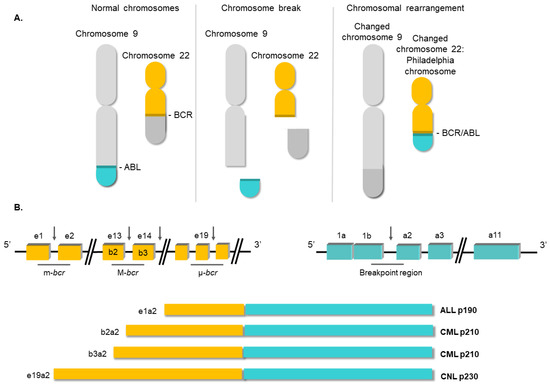
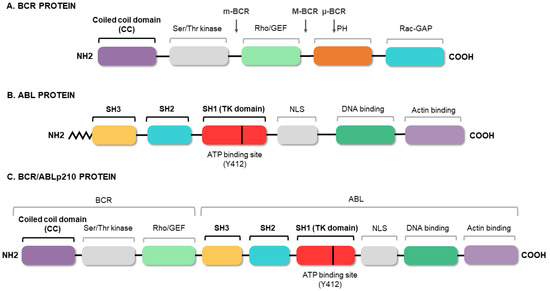
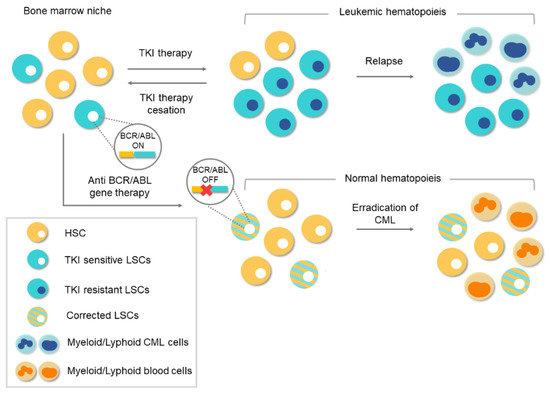
3. Conventional Therapies for Chronic Myeloid Leukemia
This entry is adapted from the peer-reviewed paper 10.3390/biology10020118
References
- Quintás-Cardama, A.; Cortes, J. Chronic Myeloid Leukemia: Diagnosis and Treatment. In Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2006; Volume 81, pp. 973–988.
- Mendizabal, A.M.; Garcia-Gonzalez, P.; Levine, P.H. Regional variations in age at diagnosis and overall survival among patients with chronic myeloid leukemia from low and middle income countries. Cancer Epidemiol. 2013, 37, 247–254.
- Petzer, A.; Eaves, C.; Lansdorp, P.; Ponchio, L.; Barnett, M.; Eaves, A. Characterization of primitive subpopulations of normal and leukemic cells present in the blood of patients with newly diagnosed as well as established chronic myeloid leukemia. Blood 1996, 88, 2162–2171.
- Melo, J.V.; Barnes, D.J. Chronic Myeloid Leukemia: Biology of Advanced Phase. In Myeloproliferative Disorders; Springer: Berlin/Heidelberg, Germany, 2007; pp. 37–58.
- Kantarjian, H.M.; Keating, M.J.; Talpaz, M.; Walters, R.S.; Smith, T.L.; Cork, A.; McCredie, K.B.; Freireich, E.J. Chronic myelogenous leukemia in blast crisis. Am. J. Med. 1987, 83, 445–454.
- Ilaria, R.L. Pathobiology of Lymphoid and Myeloid Blast Crisis and Management Issues. Hematol. Am. Soc. Hematol. Educ. Program 2005, 2005, 188–194.
- Nowell, P. A minute chromosome in human chronic granulocytic leukemia. Science 1960, 132, 1497–1499.
- Kantarjian, H.; O’Brien, S.; Jabbour, E.; Garcia-Manero, G.; Quintas-Cardama, A.; Shan, J.; Rios, M.B.; Ravandi, F.; Faderl, S.; Kadia, T.; et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: A single-institution historical experience. Blood 2012, 119, 1981–1987.
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94 (Suppl. 2), S107–S121.
- Bower, H.; Björkholm, M.; Dickman, P.W.; Höglund, M.; Lambert, P.C.; Andersson, T.M.-L. Life Expectancy of Patients with Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. J. Clin. Oncol. 2016, 34, 2851–2857.
- Deininger, M.; O’Brien, S.G.; Guilhot, F.; Goldman, D.J.M.; Hochhaus, A.; Hughes, T.P.; Radich, J.P.; Hatfield, A.K.; Mone, M.; Filian, J.; et al. International Randomized Study of Interferon Vs STI571 (IRIS) 8-Year Follow up: Sustained Survival and Low Risk for Progression or Events in Patients with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Treated with Imatinib. Blood 2009, 114, 1126.
- Graham, S.M.; Jørgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325.
- Rowley, J.D. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nat. Cell Biol. 1973, 243, 290–293.
- Ben-Neriah, Y.; Daley, G.Q.; Mes-Masson, A.M.; Witte, O.N.; Baltimore, D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986, 233, 212–214.
- Score, J.; Calasanz, M.J.; Ottman, O.; Pane, F.; Yeh, R.F.; A Sobrinho-Simões, M.; Kreil, S.; Ward, D.; Hidalgo-Curtis, C.; Melo, J.V.; et al. Analysis of genomic breakpoints in p190 and p210 BCR–ABL indicate distinct mechanisms of formation. Leukemia 2010, 24, 1742–1750.
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; De Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984, 36, 93–99.
- Verstovsek, S.; Lin, H.; Kantarjian, H.; Saglio, G.; De Micheli, D.; Pane, F.; Garcia-Manero, G.; Intrieri, M.; Rotoli, B.; Salvatore, F.; et al. Neutrophilic-chronic myeloid leukemia: Low levels of p230 BCR/ABL mRNA and undetectable p230 BCR/ABL protein may predict an indolent course. Cancer 2002, 94, 2416–2425.
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science 1990, 247, 1079–1082.
- Kabarowski, J.H.; Witte, O.N. Consequences of BCR–ABL Expression within the Hematopoietic Stem Cell in Chronic Myeloid Leukemia. Stem Cells 2000, 18, 399–408.
- Zhou, H.; Xu, R. Leukemia stem cells: The root of chronic myeloid leukemia. Protein Cell 2015, 6, 403–412.
- Janossy, G.; Roberts, M.; Greaves, M. Target Cell in Chronic Myeloid Leukæmia and Its Relationship to Acute Lymphoid Leukæmia. Lancet 1976, 308, 1058–1061.
- Cohen, G.B.; Ren, R.; Baltimore, D. Modular binding domains in signal transduction proteins. Cell 1995, 80, 237–248.
- Mayer, B.J.; Baltimore, D. Mutagenic analysis of the roles of SH2 and SH3 domains in regulation of the Abl tyrosine kinase. Mol. Cell. Biol. 1994, 14, 2883–2894.
- McWhirter, J.R.; Galasso, D.L.; Wang, J.Y. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol. Cell. Biol. 1993, 13, 7587–7595.
- Ma, G.; Lu, D.; Wu, Y.; Liu, J.; Arlinghaus, R.B. Bcr phosphorylated on tyrosine 177 binds Grb2. Oncogene 1997, 14, 2367–2372.
- Steelman, L.S.; A Franklin, R.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094.
- Walker, S.R.; Frank, D.A. STAT Signaling in the Pathogenesis and Treatment of Cancer. In Signaling Pathways in Cancer Pathogenesis and Therapy; Springer: New York, NY, USA, 2012; pp. 95–108.
- Martelli, A.M.; Evangelisti, C.; Chappell, W.; Abrams, S.L.; Bäsecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; Libra, M.; et al. Targeting the translational apparatus to improve leukemia therapy: Roles of the PI3K/PTEN/Akt/mTOR pathway. Leukemia 2011, 25, 1064–1079.
- Puil, L.; Liu, J.; Gish, G.; Mbamalu, G.; Bowtell, D.; Pelicci, P.; Arlinghaus, R.; Pawson, T. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J. 1994, 13, 764–773.
- Bedi, A.; Zehnbauer, B.; Barber, J.; Sharkis, S.; Jones, R. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood 1994, 83, 2038–2044.
- Gordon, M.Y.; Dowding, C.R.; Riley, G.; Goldman, J.M.; Greaves, M.F. Altered adhesive interactions with marrow stroma of haematopoietic progenitor cells in chronic myeloid leukaemia. Nat. Cell Biol. 1987, 328, 342–344.
- Clarkson, B.H.; Strife, A.; Wisniewski, D.; Lambek, C.; Liu, C. Chronic myelogenous leukemia as a paradigm of early cancer and possible curative strategies. Leukemia 2003, 17, 1211–1262.
- Pendergast, A.M.; Quilliam, L.A.; Cripe, L.D.; Bassing, C.H.; Dai, Z.; Li, N.; Batzer, A.; Rabun, K.M.; Der, C.J.; Schlessinger, J.; et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell 1993, 75, 175–185.
- Minot, G.R.; Buckman, T.E.; Isaacs, R. Chronic myelogenous leukemia: Age incidence, duration, and benefit derived from irradiation. J. Am. Med. Assoc. 1924, 82, 1489–1494.
- Rushing, D.; Goldman, A.; Gibbs, G.; Howe, R.; Kennedy, B.J. Hydroxyurea versus busulfan in the treatment of chronic myelogenous leukemia. Am. J. Clin. Oncol. Cancer Clin. Trials 1982, 5, 307–314.
- Tura, S.; Baccarani, M. α-interferon in the treatment of chronic myeloid leukemia. Blood 1995, 85, 2999–3000.
- Lübking, A.; Dreimane, A.; Sandin, F.; Isaksson, C.; Märkevärn, B.; Brune, M.; Ljungman, P.; Lenhoff, S.; Stenke, L.; Höglund, M.; et al. Allogeneic stem cell transplantation for chronic myeloid leukemia in the TKI era: Population-based data from the Swedish CML registry. Bone Marrow Transplant. 2019, 54, 1764–1774.
- Gale, R.P.; Hehlmann, R.; Zhang, M.J.; Hasford, J.; Goldman, J.M.; Heimpel, H.; Hochhaus, A.; Klein, J.P.; Kolb, H.J.; McGlave, P.B.; et al. Survival with Bone Marrow Transplantation Versus Hydroxyurea or Interferon for Chronic Myelogenous Leukemia. Blood 1998, 91, 1810–1819.
- Van Rhee, F.; Szydlo, R.; Hermans, J.; Devergie, A.; Frassoni, F.; Arcese, W.; De Witte, T.; Kolb, H.; Niederwiser, D.; Jacobsen, N.; et al. Long-term results after allogeneic bone marrow transplantation for chronic myelogenous leukemia in chronic phase: A report from the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 1997, 20, 553–560.
- Buchdunger, E.; Zimmermann, J.; Mett, H.; Meyer, T.; Müller, M.; Druker, B.J.; Lydon, N.B. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996, 56, 100–104.
- Bhatia, R.; A Munthe, H.; Verfaillie, C. Tyrphostin AG957, a tyrosine kinase inhibitor with anti-BCR/ABL tyrosine kinase activity restores β1 integrin-mediated adhesion and inhibitory signaling in chronic myelogenous leukemia hematopoietic progenitors. Leukemia 1998, 12, 1708–1717.
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 2002, 1, 493–502.
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N. Engl. J. Med. 2001, 344, 1031–1037.
- Milojkovic, D.; Apperley, J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 7519–7527.
- Jabbour, E.; Kantarjian, H.; Cortes, J. Use of Second- and Third-Generation Tyrosine Kinase Inhibitors in the Treatment of Chronic Myeloid Leukemia: An Evolving Treatment Paradigm. Clin. Lymphoma Myeloma Leuk. 2015, 15, 323–334.
- Mojica, F.J.M.; Montoliu, L. On the Origin of CRISPR-Cas Technology: From Prokaryotes to Mammals. Trends Microbiol. 2016, 24, 811–820.
- Wassef, M.; Luscan, A.; Battistella, A.; Le Corre, S.; Li, H.; Wallace, M.; Vidaud, M.; Margueron, R. Versatile and precise gene-targeting strategies for functional studies in mammalian cell lines. Methods 2017, 121–122, 45–54.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821.
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782.