The development of tumors requires an initiator event, usually exposure to DNA damaging agents that cause genetic alterations such as gene mutations or chromosomal abnormalities, leading to deregulated cell proliferation. Although the mere stochastic accumulation of further mutations may cause tumor progression, it is now clear that an inflammatory microenvironment has a major tumor-promoting influence on initiated cells, in particular when a chronic inflammatory reaction already existed before the initiated tumor cell was formed. Moreover, inflammatory cells become mobilized in response to signals emanating from tumor cells. In both cases, the microenvironment provides signals that initiated tumor cells perceive by membrane receptors and transduce via downstream kinase cascades to modulate multiple cellular processes and respond with changes in cell gene expression, metabolism, and morphology. Cytokines, chemokines, and growth factors are examples of major signals secreted by immune cells, fibroblast, and endothelial cells and mediate an intricate cell-cell crosstalk in an inflammatory microenvironment, which contributes to increased cancer cell survival, phenotypic plasticity and adaptation to surrounding tissue conditions. Eventually, consequent changes in extracellular matrix stiffness and architecture, coupled with additional genetic alterations, further fortify the malignant progression of tumor cells, priming them for invasion and metastasis.
- tumor microenvironment
- inflammation
- signal transduction
- cancer
1. Introduction
2. Cancer-Associated Inflammation (CAI) and the TME
2.1. Mediators of Cancer-Associated Inflammation (CAI)
2.1.1. Pro-Inflammatory Factors
2.1.2. Pro-Inflammatory Chemokines in the TME
2.1.3. Anti-Inflammatory Mediators—IL-10
2.1.4. The Transforming Growth Factor-β (TGF-β)
2.2. Signaling in the Inflammatory TME
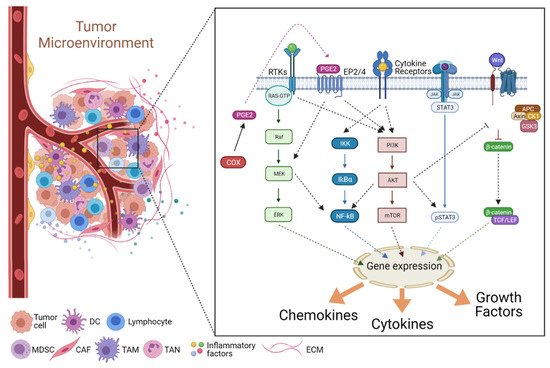
2.2.1. The JAK/STAT Pathway
2.2.2. The NF-κB Pathway
2.2.3. The COX2/PGE2 Pathway
2.2.4. The PI3K/Akt Pathway
2.2.5. The Wnt Pathway
This entry is adapted from the peer-reviewed paper 10.3390/immuno1020007
References
- Li, L.; Yu, R.; Cai, T.; Chen, Z.; Lan, M.; Zou, T.; Wang, B.; Wang, Q.; Zhao, Y.; Cai, Y. Effects of Immune Cells and Cytokines on Inflammation and Immunosuppression in the Tumor Microenvironment. Int. Immunopharmacol. 2020, 88, 106939.
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell Sci. 2012, 125, 5591–5596.
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and Chemokines: At the Crossroads of Cell Signalling and Inflammatory Disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582.
- Dinarello, C.A. The Paradox of Pro-Inflammatory Cytokines in Cancer. Cancer Metastasis Rev. 2006, 25, 307–313.
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-Related Inflammation, the Seventh Hallmark of Cancer: Links to Genetic Instability. Carcinogenesis 2009, 30, 1073–1081.
- Schenk, M.; Fabri, M.; Krutzik, S.R.; Lee, D.J.; Vu, D.M.; Sieling, P.A.; Montoya, D.; Liu, P.T.; Modlin, R.L. Interleukin-1β Triggers the Differentiation of Macrophages with Enhanced Capacity to Present Mycobacterial Antigen to T Cells. Immunology 2014, 141, 174–180.
- Apte, R.N.; Krelin, Y.; Song, X.; Dotan, S.; Recih, E.; Elkabets, M.; Carmi, Y.; Dvorkin, T.; White, R.M.; Gayvoronsky, L.; et al. Effects of Micro-Environment- and Malignant Cell-Derived Interleukin-1 in Carcinogenesis, Tumour Invasiveness and Tumour-Host Interactions. Eur. J. Cancer Oxf. Engl. 1990 2006, 42, 751–759.
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 9.
- Voronov, E.; Apte, R.N. IL-1 in Colon Inflammation, Colon Carcinogenesis and Invasiveness of Colon Cancer. Cancer Microenviron. Off. J. Int. Cancer Microenviron. Soc. 2015, 8, 187–200.
- Oh, K.; Lee, O.-Y.; Park, Y.; Seo, M.W.; Lee, D.-S. IL-1β Induces IL-6 Production and Increases Invasiveness and Estrogen-Independent Growth in a TG2-Dependent Manner in Human Breast Cancer Cells. BMC Cancer 2016, 16, 724.
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5.
- Akagi, Y.; Liu, W.; Xie, K.; Zebrowski, B.; Shaheen, R.M.; Ellis, L.M. Regulation of Vascular Endothelial Growth Factor Expression in Human Colon Cancer by Interleukin-1beta. Br. J. Cancer 1999, 80, 1506–1511.
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The Role of IL-1β in the Early Tumor Cell-Induced Angiogenic Response. J. Immunol. 2013, 190, 3500–3509.
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197.
- Lewis, A.M.; Varghese, S.; Xu, H.; Alexander, H.R. Interleukin-1 and Cancer Progression: The Emerging Role of Interleukin-1 Receptor Antagonist as a Novel Therapeutic Agent in Cancer Treatment. J. Transl. Med. 2006, 4, 48.
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 Signaling as a Promising Target to Improve the Efficacy of Cancer Immunotherapy. Cancer Sci. 2017, 108, 1947–1952.
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of Interleukin-6 in Cancer Progression and Therapeutic Resistance. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 11553–11572.
- Rossi, J.-F.; Lu, Z.-Y.; Jourdan, M.; Klein, B. Interleukin-6 as a Therapeutic Target. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1248–1257.
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 Released by Colon Cancer-Associated Fibroblasts Is Critical for Tumour Angiogenesis: Anti-Interleukin-6 Receptor Antibody Suppressed Angiogenesis and Inhibited Tumour-Stroma Interaction. Br. J. Cancer 2014, 110, 469–478.
- Gough, P.; Myles, I.A. Tumor Necrosis Factor Receptors: Pleiotropic Signaling Complexes and Their Differential Effects. Front. Immunol. 2020, 11, 585880.
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190.
- Fakhoury, M.; Negrulj, R.; Mooranian, A.; Al-Salami, H. Inflammatory Bowel Disease: Clinical Aspects and Treatments. J. Inflamm. Res. 2014, 7, 113–120.
- Pereira, R.; Faria, R.; Lago, P.; Torres, T. Infection and Malignancy Risk in Patients Treated with TNF Inhibitors for Immune-Mediated Inflammatory Diseases. Curr. Drug Saf. 2017, 12, 162–170.
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572.
- Waugh, D.J.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 6735–6741.
- Bikfalvi, A.; Billottet, C. The CC and CXC Chemokines: Major Regulators of Tumor Progression and the Tumor Microenvironment. Am. J. Physiol. Cell Physiol. 2020, 318, C542–C554.
- Wang, L.; Kuang, Z.; Zhang, D.; Gao, Y.; Ying, M.; Wang, T. Reactive Oxygen Species in Immune Cells: A New Antitumor Target. Biomed. Pharmacother. 2021, 133, 110978.
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the Interleukin-10 Receptor. Annu. Rev. Immunol. 2001, 19, 683–765.
- Zhu, L.; Shi, T.; Zhong, C.; Wang, Y.; Chang, M.; Liu, X. IL-10 and IL-10 Receptor Mutations in Very Early Onset Inflammatory Bowel Disease. Gastroenterol. Res. 2017, 10, 65–69.
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-Wide Meta-Analysis Increases to 71 the Number of Confirmed Crohn’s Disease Susceptibility Loci. Nat. Genet. 2010, 42, 1118–1125.
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 Receptor Signaling in Innate Immune Cells Regulates Mucosal Immune Tolerance and Anti-Inflammatory Macrophage Function. Immunity 2014, 40, 706–719.
- Coperchini, F.; Chiovato, L.; Ricci, G.; Croce, L.; Magri, F.; Rotondi, M. The Cytokine Storm in COVID-19: Further Advances in Our Understanding the Role of Specific Chemokines Involved. Cytokine Growth Factor Rev. 2021, 58, 82–91.
- Rossi, J.-F.; Lu, Z.Y.; Massart, C.; Levon, K. Dynamic Immune/Inflammation Precision Medicine: The Good and the Bad Inflammation in Infection and Cancer. Front. Immunol. 2021, 12, 595722.
- Cobb, D.A.; Lee, D.W. Cytokine Release Syndrome Biology and Management. Cancer J. Sudbury Mass 2021, 27, 119–125.
- Oft, M. IL-10: Master Switch from Tumor-Promoting Inflammation to Antitumor Immunity. Cancer Immunol. Res. 2014, 2, 194–199.
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230.
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev. 2018, 32, 1267–1284.
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822.
- Derynck, R.; Turley, S.J.; Akhurst, R.J. TGFβ Biology in Cancer Progression and Immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 9–34.
- Coricello, A.; Mesiti, F.; Lupia, A.; Maruca, A.; Alcaro, S. Inside Perspective of the Synthetic and Computational Toolbox of JAK Inhibitors: Recent Updates. Mol. Basel Switz. 2020, 25, 3321.
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A Multifaceted Oncoprotein. Growth Factors Chur Switz. 2018, 36, 1–14.
- Ji, Z.; He, L.; Regev, A.; Struhl, K. Inflammatory Regulatory Network Mediated by the Joint Action of NF-KB, STAT3, and AP-1 Factors Is Involved in Many Human Cancers. Proc. Natl. Acad. Sci. USA 2019, 116, 9453–9462.
- Fu, L.-Q.; Du, W.-L.; Cai, M.-H.; Yao, J.-Y.; Zhao, Y.-Y.; Mou, X.-Z. The Roles of Tumor-Associated Macrophages in Tumor Angiogenesis and Metastasis. Cell. Immunol. 2020, 353, 104119.
- Leng, K.; Xu, Y.; Kang, P.; Qin, W.; Cai, H.; Wang, H.; Ji, D.; Jiang, X.; Li, J.; Li, Z.; et al. Akirin2 Is Modulated by MiR-490-3p and Facilitates Angiogenesis in Cholangiocarcinoma through the IL-6/STAT3/VEGFA Signaling Pathway. Cell Death Dis. 2019, 10, 262.
- Yuan, S.; Zhang, S.; Zhuang, Y.; Zhang, H.; Bai, J.; Hou, Q. Interleukin-17 Stimulates STAT3-Mediated Endothelial Cell Activation for Neutrophil Recruitment. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 2340–2356.
- Wang, Y.; Chen, Q.; Zhang, Z.; Jiang, F.; Meng, X.; Yan, H. Interleukin-10 Overexpression Improves the Function of Endothelial Progenitor Cells Stimulated with TNF-α through the Activation of the STAT3 Signaling Pathway. Int. J. Mol. Med. 2015, 35, 471–477.
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF Drives Cancer-Initiating Stem Cells through VEGFR-2/Stat3 Signaling to Upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119.
- Chen, L.; Han, X. Anti-PD-1/PD-L1 Therapy of Human Cancer: Past, Present, and Future. J. Clin. Investig. 2015, 125, 3384–3391.
- Bloom, M.J.; Saksena, S.D.; Swain, G.P.; Behar, M.S.; Yankeelov, T.E.; Sorace, A.G. The Effects of IKK-Beta Inhibition on Early NF-Kappa-B Activation and Transcription of Downstream Genes. Cell. Signal. 2019, 55, 17–25.
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.-W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta Links Inflammation and Tumorigenesis in a Mouse Model of Colitis-Associated Cancer. Cell 2004, 118, 285–296.
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-KappaB in Cancer: From Innocent Bystander to Major Culprit. Nat. Rev. Cancer 2002, 2, 301–310.
- Aggarwal, B.B. Nuclear Factor-KappaB: The Enemy Within. Cancer Cell 2004, 6, 203–208.
- Lalle, G.; Twardowski, J.; Grinberg-Bleyer, Y. NF-ΚB in Cancer Immunity: Friend or Foe? Cells 2021, 10, 355.
- Fan, Y.; Mao, R.; Yang, J. NF-ΚB and STAT3 Signaling Pathways Collaboratively Link Inflammation to Cancer. Protein Cell 2013, 4, 176–185.
- Taniguchi, K.; Karin, M. NF-ΚB, Inflammation, Immunity and Cancer: Coming of Age. Nat. Rev. Immunol. 2018, 18, 309–324.
- Papila, K.B.; Sozer, V.; Cigdem, K.P.; Durmus, S.; Kurtulus, D.; Papila, C.; Gelisgen, R.; Uzun, H. Circulating Nuclear Factor-Kappa B Mediates Cancer-Associated Inflammation in Human Breast and Colon Cancer. J. Med. Biochem. 2021, 40, 150–159.
- Dong, F.; Zhou, X.; Li, C.; Yan, S.; Deng, X.; Cao, Z.; Li, L.; Tang, B.; Allen, T.D.; Liu, J. Dihydroartemisinin Targets VEGFR2 via the NF-ΚB Pathway in Endothelial Cells to Inhibit Angiogenesis. Cancer Biol. Ther. 2014, 15, 1479–1488.
- Martin, D.; Galisteo, R.; Gutkind, J.S. CXCL8/IL8 Stimulates Vascular Endothelial Growth Factor (VEGF) Expression and the Autocrine Activation of VEGFR2 in Endothelial Cells by Activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) Complex. J. Biol. Chem. 2009, 284, 6038–6042.
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting Nuclear Factor-Kappa B to Overcome Resistance to Chemotherapy. Front. Oncol. 2013, 3, 120.
- De, D.; Chowdhury, P.; Panda, S.K.; Ghosh, U. Ethanolic Extract of Leaf of Dillenia Pentagyna Reduces In-Vitro Cell Migration and Induces Intrinsic Pathway of Apoptosis via Downregulation of NF-Κβ in Human NSCLC A549 Cells. J. Cell. Biochem. 2019, 120, 19841–19857.
- Huang, L.; Jian, Z.; Gao, Y.; Zhou, P.; Zhang, G.; Jiang, B.; Lv, Y. RPN2 Promotes Metastasis of Hepatocellular Carcinoma Cell and Inhibits Autophagy via STAT3 and NF-ΚB Pathways. Aging 2019, 11, 6674–6690.
- Pu, D.; Yin, L.; Huang, L.; Qin, C.; Zhou, Y.; Wu, Q.; Li, Y.; Zhou, Q.; Li, L. Cyclooxygenase-2 Inhibitor: A Potential Combination Strategy With Immunotherapy in Cancer. Front. Oncol. 2021, 11, 637504.
- Wang, D.; Dubois, R.N. Eicosanoids and Cancer. Nat. Rev. Cancer 2010, 10, 181–193.
- Ferrer, M.D.; Busquets-Cortés, C.; Capó, X.; Tejada, S.; Tur, J.A.; Pons, A.; Sureda, A. Cyclooxygenase-2 Inhibitors as a Therapeutic Target in Inflammatory Diseases. Curr. Med. Chem. 2019, 26, 3225–3241.
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270.
- Pan, Q.; Bao, L.W.; Merajver, S.D. Tetrathiomolybdate Inhibits Angiogenesis and Metastasis through Suppression of the NFkappaB Signaling Cascade. Mol. Cancer Res. MCR 2003, 1, 701–706.
- Wang, D.; DuBois, R.N. Immunosuppression Associated with Chronic Inflammation in the Tumor Microenvironment. Carcinogenesis 2015, 36, 1085–1093.
- Escuin-Ordinas, H.; Atefi, M.; Fu, Y.; Cass, A.; Ng, C.; Huang, R.R.; Yashar, S.; Comin-Anduix, B.; Avramis, E.; Cochran, A.J.; et al. COX-2 Inhibition Prevents the Appearance of Cutaneous Squamous Cell Carcinomas Accelerated by BRAF Inhibitors. Mol. Oncol. 2014, 8, 250–260.
- Panza, E.; De Cicco, P.; Ercolano, G.; Armogida, C.; Scognamiglio, G.; Anniciello, A.M.; Botti, G.; Cirino, G.; Ianaro, A. Differential Expression of Cyclooxygenase-2 in Metastatic Melanoma Affects Progression Free Survival. Oncotarget 2016, 7, 57077–57085.
- Miao, J.; Lu, X.; Hu, Y.; Piao, C.; Wu, X.; Liu, X.; Huang, C.; Wang, Y.; Li, D.; Liu, J. Prostaglandin E2 and PD-1 Mediated Inhibition of Antitumor CTL Responses in the Human Tumor Microenvironment. Oncotarget 2017, 8, 89802–89810.
- Gu, K.J.; Li, G. An Overview of Cancer Prevention: Chemoprevention and Immunoprevention. J. Cancer Prev. 2020, 25, 127–135.
- Markosyan, N.; Li, J.; Sun, Y.H.; Richman, L.P.; Lin, J.H.; Yan, F.; Quinones, L.; Sela, Y.; Yamazoe, T.; Gordon, N.; et al. Tumor Cell-Intrinsic EPHA2 Suppresses Anti-Tumor Immunity by Regulating PTGS2 (COX-2). J. Clin. Investig. 2019, 129, 3594–3609.
- Yan, G.; Zhao, H.; Zhang, Q.; Zhou, Y.; Wu, L.; Lei, J.; Wang, X.; Zhang, J.; Zhang, X.; Zheng, L.; et al. A RIPK3-PGE2 Circuit Mediates Myeloid-Derived Suppressor Cell-Potentiated Colorectal Carcinogenesis. Cancer Res. 2018, 78, 5586–5599.
- Li, A.; Chen, P.; Leng, Y.; Kang, J. Histone Deacetylase 6 Regulates the Immunosuppressive Properties of Cancer-Associated Fibroblasts in Breast Cancer through the STAT3-COX2-Dependent Pathway. Oncogene 2018, 37, 5952–5966.
- Chiang, K.-H.; Shieh, J.-M.; Shen, C.-J.; Chang, T.-W.; Wu, P.-T.; Hsu, J.-Y.; Tsai, J.-P.; Chang, W.-C.; Chen, B.-K. Epidermal Growth Factor-Induced COX-2 Regulates Metastasis of Head and Neck Squamous Cell Carcinoma through Upregulation of Angiopoietin-like 4. Cancer Sci. 2020, 111, 2004–2015.
- Smakman, N.; Kranenburg, O.; Vogten, J.M.; Bloemendaal, A.L.A.; van Diest, P.; Borel Rinkes, I.H.M. Cyclooxygenase-2 Is a Target of KRASD12, Which Facilitates the Outgrowth of Murine C26 Colorectal Liver Metastases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 41–48.
- Lim, H.J.; Park, J.H.; Jo, C.; Yoon, K.; Koh, Y.H. Cigarette Smoke Extracts and Cadmium Induce COX-2 Expression through γ-Secretase-Mediated P38 MAPK Activation in C6 Astroglia Cells. PLoS ONE 2019, 14, e0212749.
- Tang, F.; Wang, Y.; Hemmings, B.A.; Rüegg, C.; Xue, G. PKB/Akt-Dependent Regulation of Inflammation in Cancer. Semin. Cancer Biol. 2018, 48, 62–69.
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The Relation between PI3K/AKT Signalling Pathway and Cancer. Gene 2019, 698, 120–128.
- Xu, K.; Liu, P.; Wei, W. MTOR Signaling in Tumorigenesis. Biochim. Biophys. Acta 2014, 1846, 638–654.
- Tang, H.; Massi, D.; Hemmings, B.A.; Mandalà, M.; Hu, Z.; Wicki, A.; Xue, G. AKT-Ions with a TWIST between EMT and MET. Oncotarget 2016, 7, 62767–62777.
- Chen, Y.; Kijlstra, A.; Chen, Y.; Yang, P. IL-17A Stimulates the Production of Inflammatory Mediators via Erk1/2, P38 MAPK, PI3K/Akt, and NF-ΚB Pathways in ARPE-19 Cells. Mol. Vis. 2011, 17, 3072–3077.
- Dan, H.C.; Cooper, M.J.; Cogswell, P.C.; Duncan, J.A.; Ting, J.P.-Y.; Baldwin, A.S. Akt-Dependent Regulation of NF-B Is Controlled by MTOR and Raptor in Association with IKK. Genes Dev. 2008, 22, 1490–1500.
- Factor, V.; Oliver, A.L.; Panta, G.R.; Thorgeirsson, S.S.; Sonenshein, G.E.; Arsura, M. Roles of Akt/PKB and IKK Complex in Constitutive Induction of NF-KappaB in Hepatocellular Carcinomas of Transforming Growth Factor Alpha/c-Myc Transgenic Mice. Hepatol. Baltim. Md 2001, 34, 32–41.
- Gu, F.-M.; Li, Q.-L.; Gao, Q.; Jiang, J.-H.; Zhu, K.; Huang, X.-Y.; Pan, J.-F.; Yan, J.; Hu, J.-H.; Wang, Z.; et al. IL-17 Induces AKT-Dependent IL-6/JAK2/STAT3 Activation and Tumor Progression in Hepatocellular Carcinoma. Mol. Cancer 2011, 10, 150.
- Xue, G.; Zippelius, A.; Wicki, A.; Mandalà, M.; Tang, F.; Massi, D.; Hemmings, B.A. Integrated Akt/PKB Signaling in Immunomodulation and Its Potential Role in Cancer Immunotherapy. J. Natl. Cancer Inst. 2015, 107.
- Liu, L.-Z.; Hu, X.-W.; Xia, C.; He, J.; Zhou, Q.; Shi, X.; Fang, J.; Jiang, B.-H. Reactive Oxygen Species Regulate Epidermal Growth Factor-Induced Vascular Endothelial Growth Factor and Hypoxia-Inducible Factor-1alpha Expression through Activation of AKT and P70S6K1 in Human Ovarian Cancer Cells. Free Radic. Biol. Med. 2006, 41, 1521–1533.
- Madge, L.A.; Pober, J.S. A Phosphatidylinositol 3-Kinase/Akt Pathway, Activated by Tumor Necrosis Factor or Interleukin-1, Inhibits Apoptosis but Does Not Activate NFkappaB in Human Endothelial Cells. J. Biol. Chem. 2000, 275, 15458–15465.
- Liu, S.; Shen, H.; Xu, M.; Liu, O.; Zhao, L.; Liu, S.; Guo, Z.; Du, J. FRP Inhibits Ox-LDL-Induced Endothelial Cell Apoptosis through an Akt-NF-B-Bcl-2 Pathway and Inhibits Endothelial Cell Apoptosis in an ApoE-Knockout Mouse Model. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E351–E363.
- Massacesi, C.; Di Tomaso, E.; Urban, P.; Germa, C.; Quadt, C.; Trandafir, L.; Aimone, P.; Fretault, N.; Dharan, B.; Tavorath, R.; et al. PI3K Inhibitors as New Cancer Therapeutics: Implications for Clinical Trial Design. OncoTargets Ther. 2016, 9, 203–210.
- Suvarna, V.; Murahari, M.; Khan, T.; Chaubey, P.; Sangave, P. Phytochemicals and PI3K Inhibitors in Cancer-An Insight. Front. Pharmacol. 2017, 8, 916.
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464.
- Niehrs, C. The Complex World of WNT Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779.
- Azbazdar, Y.; Karabicici, M.; Erdal, E.; Ozhan, G. Regulation of Wnt Signaling Pathways at the Plasma Membrane and Their Misregulation in Cancer. Front. Cell Dev. Biol. 2021, 9, 631623.
- Caspi, M.; Wittenstein, A.; Kazelnik, M.; Shor-Nareznoy, Y.; Rosin-Arbesfeld, R. Therapeutic Targeting of the Oncogenic Wnt Signaling Pathway for Treating Colorectal Cancer and Other Colonic Disorders. Adv. Drug Deliv. Rev. 2021, 169, 118–136.
- Li, X.; Ortiz, M.A.; Kotula, L. The Physiological Role of Wnt Pathway in Normal Development and Cancer. Exp. Biol. Med. Maywood NJ 2020, 245, 411–426.
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and Mechanisms of WNT Pathway Tumour Suppressors in Cancer. Nat. Rev. Cancer 2021, 21, 5–21.
- Rasola, A.; Fassetta, M.; De Bacco, F.; D’Alessandro, L.; Gramaglia, D.; Di Renzo, M.F.; Comoglio, P.M. A Positive Feedback Loop between Hepatocyte Growth Factor Receptor and Beta-Catenin Sustains Colorectal Cancer Cell Invasive Growth. Oncogene 2007, 26, 1078–1087.
- Smith, K.; Bui, T.D.; Poulsom, R.; Kaklamanis, L.; Williams, G.; Harris, A.L. Up-Regulation of Macrophage Wnt Gene Expression in Adenoma-Carcinoma Progression of Human Colorectal Cancer. Br. J. Cancer 1999, 81, 496–502.
- Ojalvo, L.S.; Whittaker, C.A.; Condeelis, J.S.; Pollard, J.W. Gene Expression Analysis of Macrophages That Facilitate Tumor Invasion Supports a Role for Wnt-Signaling in Mediating Their Activity in Primary Mammary Tumors. J. Immunol. 2010, 184, 702–712.
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 Promotes Colon Cancer Cell Growth through a Gs-Axin-Beta-Catenin Signaling Axis. Science 2005, 310, 1504–1510.
- Lopez-Bergami, P.; Barbero, G. The Emerging Role of Wnt5a in the Promotion of a Pro-Inflammatory and Immunosuppressive Tumor Microenvironment. Cancer Metastasis Rev. 2020, 39, 933–952.
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79.
- Pereira, C.; Schaer, D.J.; Bachli, E.B.; Kurrer, M.O.; Schoedon, G. Wnt5A/CaMKII Signaling Contributes to the Inflammatory Response of Macrophages and Is a Target for the Antiinflammatory Action of Activated Protein C and Interleukin-10. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 504–510.
- Ge, X.-P.; Gan, Y.-H.; Zhang, C.-G.; Zhou, C.-Y.; Ma, K.-T.; Meng, J.-H.; Ma, X.-C. Requirement of the NF-ΚB Pathway for Induction of Wnt-5A by Interleukin-1β in Condylar Chondrocytes of the Temporomandibular Joint: Functional Crosstalk between the Wnt-5A and NF-ΚB Signaling Pathways. Osteoarthr. Cartil. 2011, 19, 111–117.
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Pérez-Hernández, A.I.; Gurbindo, J.; Ramírez, B.; Méndez-Giménez, L.; Rotellar, F.; Valentí, V.; Moncada, R.; et al. Activation of Noncanonical Wnt Signaling through WNT5A in Visceral Adipose Tissue of Obese Subjects Is Related to Inflammation. J. Clin. Endocrinol. Metab. 2014, 99, E1407–E1417.
- Park, S.-Y.; Kang, M.-J.; Han, J.-S. Interleukin-1 Beta Promotes Neuronal Differentiation through the Wnt5a/RhoA/JNK Pathway in Cortical Neural Precursor Cells. Mol. Brain 2018, 11, 39.
- Linnskog, R.; Mohapatra, P.; Moradi, F.; Prasad, C.P.; Andersson, T. Demonstration of a WNT5A-IL-6 Positive Feedback Loop in Melanoma Cells: Dual Interference of This Loop More Effectively Impairs Melanoma Cell Invasion. Oncotarget 2016, 7, 37790–37802.
- Rozovski, U.; Harris, D.M.; Li, P.; Liu, Z.; Jain, P.; Ferrajoli, A.; Burger, J.A.; Bose, P.; Thompson, P.A.; Jain, N.; et al. STAT3-Induced Wnt5a Provides Chronic Lymphocytic Leukemia Cells with Survival Advantage. J. Immunol. 2019, 203, 3078–3085.
- Liu, Q.; Yang, C.; Wang, S.; Shi, D.; Wei, C.; Song, J.; Lin, X.; Dou, R.; Bai, J.; Xiang, Z.; et al. Wnt5a-Induced M2 Polarization of Tumor-Associated Macrophages via IL-10 Promotes Colorectal Cancer Progression. Cell Commun. Signal. CCS 2020, 18, 51.
- Chen, Y.; Chen, L.; Yu, J.; Ghia, E.M.; Choi, M.Y.; Zhang, L.; Zhang, S.; Sanchez-Lopez, E.; Widhopf, G.F.; Messer, K.; et al. Cirmtuzumab Blocks Wnt5a/ROR1 Stimulation of NF-ΚB to Repress Autocrine STAT3 Activation in Chronic Lymphocytic Leukemia. Blood 2019, 134, 1084–1094.
- Barbero, G.; Castro, M.V.; Villanueva, M.B.; Quezada, M.J.; Fernández, N.B.; DeMorrow, S.; Lopez-Bergami, P. An Autocrine Wnt5a Loop Promotes NF-ΚB Pathway Activation and Cytokine/Chemokine Secretion in Melanoma. Cells 2019, 8, 1060.
- Ikeda, T.; Nishita, M.; Hoshi, K.; Honda, T.; Kakeji, Y.; Minami, Y. Mesenchymal Stem Cell-Derived CXCL16 Promotes Progression of Gastric Cancer Cells by STAT3-Mediated Expression of Ror1. Cancer Sci. 2020, 111, 1254–1265.