The therapeutic approach to Chronic Myeloid Leukemia (CML) has changed since the advent of the tyrosine kinase inhibitor (TKI) imatinib, which was then followed by the second generation TKIs dasatinib, nilotinib, and, finally, by ponatinib, a third-generation drug. At present, these therapeutic options represent the first-line treatment for adults. Based on clinical experience, imatinb, dasatinib, and nilotinib have been approved for children even though the studies that were concerned with efficacy and safety toward pediatric patients are still awaiting more specific and high-quality data.
- chronic myeloid leukemia
- tyrosine kinase inhibitors
- pediatric age
- imatinib
- dasatinib
- nilotinb
- ponatinib
Definition
Chronic myeloid leukemia (CML) is a myeloproliferative disorder, characterized by an abnormal granulocyte cells proliferation determining a high increase of white blood cell count in addition to a spleen enlargement (splenomegaly). The CML pathogenesis stems from the Philadelphia chromosome, discovered by Peter Noweel in 1960. It arises from a translocation of the TK ABL (Abelson) gene from chromosome 9 to chromosome 22, on the BCR gene (breakpoint cluster region). The translocation generates the oncogenic BCR-ABL1 fusion gene in hematopoietic stem cells, which encodes an abnormal protein, with constitutive TK activity, responsible for proliferative and anti-apoptotic signals. The occurrence of BCR-ABL protein kinases was observed in more than 90% of CML patients[1].
Introduction
CML has three clinical phases. The first is the chronic phase (CP) without subjective symptoms after 3–5 years from diagnosis but a high white blood cell and platelet count. The second is the accelerated phase (AP) with an incremented differentiation of abnormal granulocytes. The third phase consists in the blast crisis (BC) with an increase of undifferentiated blasts. Patients in the chronic phase can be treated with tyrosine kinases inhibitors (TKIs). Unfortunately, accelerated and blast phases are not responsive to TKIs likely because their progression is not affected by BCR-ABL.
CML Therapeutic Approach
In the past decades, the first therapeutic approach to CML involved the use of ordinary chemotherapeutical drugs (such as a busulfan, hydroxyurea, cyclophosphamide, and vincristine), which is followed by allogeneic hematopoietic stem cells transplantation (allo-HSCT). This is considered a potentially curative procedure for a variety of hematological malignancies in order to reconstitute hematopoiesis [2].
However, allo-HSCT is limited by the availability of donors, transplant-related mortality, and early morbidity, so that it is considered a third-line treatment for most pediatric cases. Therefore, Bcr-Abl inhibitors may be accounted as a targeted therapy toward CML, avoiding all the side effects associated with canonical chemotherapy, and graft-versus-host disease, which is one of the major complications of allogeneic hemapoietic cell transplantation (HCT).
Currently, Imatinib is not the only TK inhibitor approved for patients with CML-CP (CML-Chronic Phase) including pediatric patients, but it remains the first-line therapy for patients with CML-AP (CML-Accellerate Phase) and CML-BC (CML-Blast Crisis).
Imatinib (Figure 1A) was discovered in 1992 as a selective inhibitor of Bcr-Abl TK. It is able to link to the kinase in its inactive form preventing the ATP binding (Figure 1A). It was approved in 2001 by FDA as first-line therapy for CML patients by obtaining optimal results compared to those who had been treated with hydroxyurea and IFN-α in terms of CHR (Complete Hematological Response), CCR (Complete Cytogenetic Response), and MMR (Major Molecular Response). Unfortunately, the appearance of resistance phenomena required the development of a second generation of TKI [3].
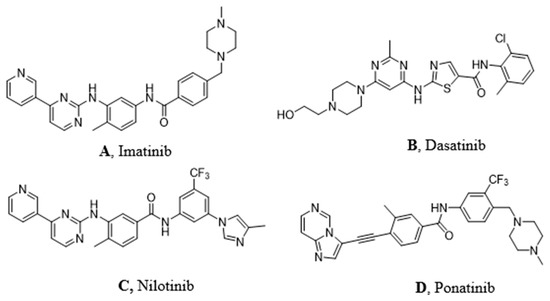
Figure 1. Molecular structures of the principal BCR-ABL TKIs used for treating pediatric chronic myeloid leukemia (CML): (A) Imatinib; (B) Dasatinib; (C) Nilotinib; (D) Ponatinib.
Resistance and Intolerance to Imatinib
The urgent need to develop a second-generation of TK inhibitors was principally due to the resistance and the intolerance of some patients to imatinib. A patient can be defined resistant to imatinib when presenting an increasing white blood cell or platelets counts that prove a primary resistance or a hematologic relapse. Moreover, resistance to imatinib consists of suboptimal cytogenetic or molecular response or failure, progression to AP/BC, reappearance of Ph+ bone marrow cells following achievement of complete cytogenetic response (CCR) and, at least, increase of more than 30% in Ph+ cells in peripheral blood or bone marrow, or loss of a molecular response. Frequently, resistance is associated with emerging mutations in some BCR-ABL amino acid residues. We can count 12 residues with risk of mutations that can be classified in resistant (T315I), less sensitive (Y235H, F359V, E255K), and sensitive (all other mutations except those mentioned before).[4]
Other resistance phenomena such as P-gp (Glycoprotein P) efflux pump overexpression, OCT1 (Organic Cation transporter) reduced expression, and th[5]e activation of alternating oncogenic pathways (Src (proto oncogene Sarcome tyrosin kinase), PI3K (Phosphoinositide 3-kinases), PDGFR, KRAS (Kirsten rat sarcoma Kinase), and JAK2) must be reminded [6].
Furthermore, intolerance is related to imatinib side effects due to its off-target activity. Among all, we can remember the increase of blood bilirubin, headache, rash, vomiting, cardiotoxicity, neutropenia, obesity, and abdominal pain.[7]
A patient can be defined intolerant to imatinib only when he develops serious adverse events (AEs) such as renal failure or liver injury (hepatic transaminase and bilirubin elevations), which require discontinuation or change of therapy.[8]
Second Generation TKI
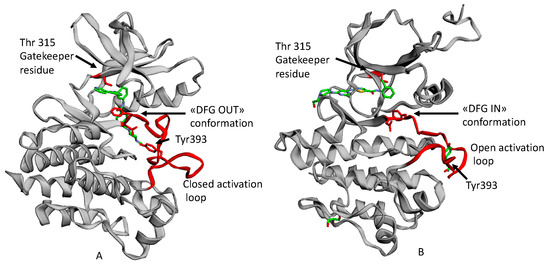
Dasatinib (Figure 1B) is the first second generation TKI approved by FDA in 2006. It is able to inhibit BCR-ABL both in the DGF in (Figure 2B) and in the DGF out conformation and also other TK such as PDGFR and Src [12]. Based on X-ray analysis, it was observed that dasatinib binds the ABL protein in a less strict conformation compared to imatinib. Despite the lower number of binding interactions observed for dasatinib vs. ABL with respect to that occurring between imatinib vs. ABL. The former is provided with inhibiting activity 325-fold higher and it is able to overcome many BCR-ABL mutations resistant to imatinib with the exception of the T315I one [9]. Furthermore, dasatinb is not P-gp substrate and, thus, can be used in patients resistant to imatinb and nilotinib.
Nilotinib (Figure 1C) is another second-generation TKI. It is a BCR-ABL, c-kit, and PDGFR inhibitor from 10-fold to 30-fold more powerful than imatinib on the BCR-ABL oncoprotein. It inhibits the proliferation of cells exhibiting the mutated TK. Nilotinib binds the protein in its inactive conformation (DFG OUT) through interactions similar to those observed for imatinib, such as four hydrogen bonds and many Van der Waals contacts. The successful introduction of the trifluoromethyl group on the phenyl ring and the substitution of the methyl-piperazine with imidazole are responsible for its greater activity. It can also overcome some Bcr-Abl mutations (L248V, G250E, Q252H) apart from T315I [10]. According to Redaelli et al.,[11] by comparing the nilotinib activity vs. imatinib and dasatinib, the former showed a mild resistance to 13 out of 18 mutations of BCR-ABL with high resistance to T315I and E255V changes. Based on some clinical trials of phase III in 2011 (ENESTnd Study), a major complete cytogenetic remission (CCR) was observed and a reduction in the percentage of patients proceeded to the accelerated phase of CML after an annual treatment with nilotinib in comparison with imatinib. For these reasons, nilotinib was approved as first-line therapy for newly diagnosed patients with CML in 2011.
Ponatinib (Figure 1D) is a very powerful third-generation pan-inhibitor of TKs. It was synthetized as a ligand of ATP binding domain of BCR-ABL in closed conformation. Ponatinib binds the DFG OUT conformation of the protein. The peculiarity of this molecule lies in the occurrence of a triple bond, which allows us to mitigate the steric hindrance due T315I mutation. The triple bond between the phenyl and the heterocycle ring in ponatinib can hold the isoleucine side chain. Having said that, ponatinib can be considered as a TK inhibitor unaffected from T315I mutation. Its binding is ensured by five hydrogen bonds. Moreover, there are several Van der Waals interactions supporting affinity in the case of multiple mutations.[12] Notably, ponatinib kept its activity in other CML mutations, such as M244V, G250E, and Q252H. However, its use drew attention to the appearance of an important side effect, which is the inhibition of parental cells Ba/F3 proliferation [13].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21124469
References
- J E Cortes; M Talpaz; Miloslav Beran; S M O'brien; M B Rios; S Stass; H M Kantarjian; Philadelphia chromosome-negative chronic myelogenous leukemia with rearrangement of the breakpoint cluster region. Long-term follow-up results.. Cancer 1995, 75, 464-470, .
- Boglarka Gyurkocza; Andrew Rezvani; Rainer F Storb; Allogeneic hematopoietic cell transplantation: the state of the art. Expert Review of Hematology 2010, 3, 285-299, 10.1586/ehm.10.21.
- Moshe Talpaz; Neil P. Shah; Hagop Kantarjian; Nicholas Donato; John Nicoll; Ron Paquette; Jorge Cortes; Susan O'brien; Claude Nicaise; Eric Bleickardt; et al. Dasatinib in Imatinib-Resistant Philadelphia Chromosome–Positive Leukemias. New England Journal of Medicine 2006, 354, 2531-2541, 10.1056/nejmoa055229.
- Valentina Nardi; Mohammad Azam; George Q. Daley; Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Current Opinion in Hematology 2004, 11, 35-43, 10.1097/00062752-200401000-00006.
- E. Premkumar Reddy; Aneel K. Aggarwal; The Ins and Outs of Bcr-Abl Inhibition. Genes & Cancer 2012, 3, 447-454, 10.1177/1947601912462126.
- Nicolas Widmer; Sara Colombo; Thierry Buclin; Laurent Arthur Decosterd; Functional consequence of MDR1 expression on imatinib intracellular concentrations. Blood 2003, 102, 1142-1142, 10.1182/blood-2003-03-0993.
- Maria Maddalena Cavalluzzi; Paola Imbrici; Roberta Gualdani; Angela Stefanachi; Giuseppe Felice Mangiatordi; Giovanni Lentini; Orazio Nicolotti; Human ether-à-go-go-related potassium channel: exploring SAR to improve drug design. Drug Discovery Today 2020, 25, 344-366, 10.1016/j.drudis.2019.11.005.
- Jorge Luis Ángeles-Velázquez; Rafael Hurtado-Monroy; Pablo Vargas-Viveros; Silvia Carrillo-Muñoz; Myrna Candelaria-Hernández; Imatinib Intolerance Is Associated With Blastic Phase Development in Philadelphia Chromosome–Positive Chronic Myeloid Leukemia. Clinical Lymphoma Myeloma and Leukemia 2016, 16, S82-S85, 10.1016/j.clml.2016.02.028.
- J. S. Tokarski; The Structure of Dasatinib (BMS-354825) Bound to Activated ABL Kinase Domain Elucidates Its Inhibitory Activity against Imatinib-Resistant ABL Mutants. Cancer Research 2006, 66, 5790-5797, 10.1158/0008-5472.can-05-4187.
- Prithviraj Bose; Haeseong Park; Jawad Al-Khafaji; Steven Grant; Strategies to circumvent the T315I gatekeeper mutation in the Bcr-Abl tyrosine kinase. Leukemia Research Reports 2013, 2, 18-20, 10.1016/j.lrr.2013.02.001.
- Sara Redaelli; Rocco Piazza; Roberta Rostagno; Vera Magistroni; Pietro Perini; Manuela Marega; Carlo B Gambacorti-Passerini; Frank Boschelli; Activity of Bosutinib, Dasatinib, and Nilotinib Against 18 Imatinib-Resistant BCR/ABL Mutants. Journal of Clinical Oncology 2009, 27, 469-471, 10.1200/jco.2008.19.8853.
- Thomas O'hare; William C. Shakespeare; Xiaotian Zhu; Christopher A. Eide; Victor M. Rivera; Frank Wang; Lauren T. Adrian; Tianjun Zhou; Wei-Sheng Huang; Qihong Xu; et al. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell 2009, 16, 401-412, 10.1016/j.ccr.2009.09.028.
- Mohammad Azam; Valentina Nardi; William C. Shakespeare; Chester A. Metcalf; Regine S. Bohacek; Yihan Wang; Raji Sundaramoorthi; Piotr Sliz; Darren R. Veach; William G. Bornmann; et al. Activity of dual SRC-ABL inhibitors highlights the role of BCR/ABL kinase dynamics in drug resistance. Proceedings of the National Academy of Sciences 2006, 103, 9244-9249, 10.1073/pnas.0600001103.