N-methyl-D-aspartate (NMDA) receptor antagonists such as phencyclidine (PCP), dizocilpine (MK-801) and ketamine have long been considered a model of schizophrenia, both in animals and humans. However, ketamine has been recently approved for treatment-resistant depression, although with severe restrictions. Interestingly, the dosage in both conditions is similar, and positive symptoms of schizophrenia appear before antidepressant effects emerge. Here, we describe the temporal mechanisms implicated in schizophrenia-like and antidepressant-like effects of NMDA blockade in rats, and postulate that such effects may indicate that NMDA receptor antagonists induce similar mechanistic effects, and only the basal pre-drug state of the organism delimitates the overall outcome.
1. Introduction
The N-Methyl-D-aspartate (NMDA) receptor (NMDAR) is an ionotropic glutamate receptor that possesses unique characteristics. The flow of ions through the channel is blocked by Mg
2+. Two different processes are necessary for activating NMDARs. First, the previous membrane depolarization removes Mg
2+ ions, and second, the additional binding of co-agonists glycine and glutamate allows voltage-dependent inflow of Na
+ and Ca
2+ ions and the outflow of K
+ ions. This dual gating by ligand binding and membrane depolarization makes the NMDAR receptor optimally fitted to function as a coincidence detector [
1]. NMDARs are involved in several physiologic functions, and their correct operation is crucial for cellular homeostasis. Any disruption in their function is thus susceptible of resulting in the manifestation of neuropsychiatric or neurological pathologies. NMDARs are critical for neuroplasticity, i.e., the ability of the brain to adapt to novel conditions. The function of NMDARs usually declines with age, which most likely contributes to the reduced plasticity that leads to learning and memory impairment. For this reason, the impairment of learning and memory seen in a variety of different pathologies, such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Huntington’s disease, Parkinson’s disease (PD), schizophrenia and major depressive disorder (MDD) are associated with NMDAR malfunction. Due to the important implication of neuronal plasticity [
2,
3], the present review is focused on the link between NMDARs and the pathophysiology and treatment of schizophrenia and depression. Two of the most important mechanisms of synaptic plasticity that are dependent on NMDAR stimulation are long-term potentiation (LTP) and long-term depression (LTD). In LTP, a high-frequency stimulation of NMDARs produces a long-lasting increase in signal transmission between two neurons [
4]. On the other hand, repetitive, low-frequency stimulation induces LTD by weakening specific synapses, which would counterbalance synaptic strengthening caused by LTP [
5].
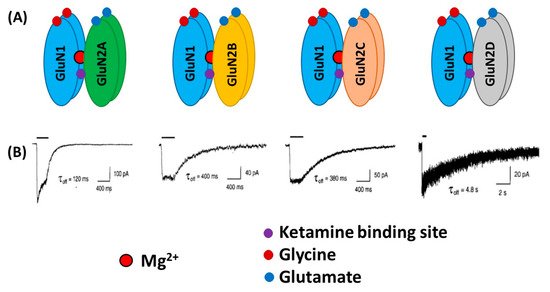
From a structural viewpoint, NMDARs are ionotropic glutamate receptors made up of four subunits. There are three different families of NMDAR subunits, i.e., GluN1, GluN2 and GluN3 (). In addition, GluN2 subunits are subdivided into GluN2A, GluN2B, GluN2C and GluN2D subunits and GluN3 subunit into GluN3A and GluN3B subunits. The ion channel of the NMDAR is formed by two necessary GluN1 subunits, and either two GluN2 subunits or a combination of GluN2 and GluN3 subunits [
6,
7,
8]. GluN1 subunits carry recognition sites for glycine, whereas GluN2 subunits possess recognition sites for glutamate, which determines the duration of channel opening and desensitization processes.
Figure 1. Schematic illustration of the N-Methyl-D-aspartate (NMDA) receptors (NMDARs) containing GluN1 and different GluN2 subtypes (A). Lower traces (B) indicate whole-cell patch-clamp recordings of responses from brief application of glutamate (1 ms of 1 mM glutamate) to recombinant diheteromeric NMDA receptor subtypes expressed in HEK293 cells. Averaged offset decay constant values (τoff) are listed below current traces. (B) “Reprinted from Neuron, Vol 12, number 3, H. Monyer, N Burnashev, D.J. Laurie, B. Sakmann, P.H. Seeburg, Developmental and regional expression in the rat brain and functional properties of four NMDA receptors, Pages No. 529-524, Copyright (1994), with permission from Elsevier”.
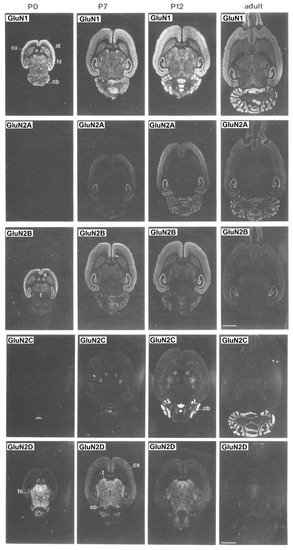
Overall, subunit composition of NMDARs changes along development and varies in different brain regions, which might influence the direction of synaptic plasticity. As depicted in , the four glutamate-binding GluN2A-D subunits, in addition to the obligatory GluN1 subunit, are the most prominent subunits in the central nervous system (CNS) [
9]. Cortical, hippocampal and striatal neurons in rodents are enriched in GluN2A and GluN2B subunits [
8,
10,
11]. The GluN2D subunit is also present in the hippocampus, but only in younger rats, being undetectable in the adulthood [
8]. In contrast, GluN2C subunits are practically restricted to cerebellum with low levels of expression in retrosplenial cortex and thalamus [
8,
12]. NMDARs are found mainly postsynaptically, although an important subset of them is also found extrasynaptically. The activation of synaptic NMDARs generally promotes synaptic and cell survival, whereas overactivation of extrasynaptic NMDARs by an excess of glutamate can be neurotoxic and induce cell death [
13]. It has been reported that GluN2A subunits are predominant at the synapses, whereas GluN2B and GluN2D are localized, though not exclusively, to extrasynaptic compartment [
14,
15,
16,
17]. Thus, GluN2A-containing receptors have been reported to contribute to synaptic plasticity, whereas GluN2B-selective antagonists may possess neuroprotective properties.
Figure 2. Distribution of the GluN1, GluN2A, GluN2B, GluN2C and GluN2D receptor subunit mRNAs. Postnatal developmental profiles of transcripts in horizontal rat brain sections from P0, P7, P12, and adult rats. Abbreviations: cb, cerebellum; cx, cortex; hi, hippocampus; s, septum; st, striatum; t, thalamus. Bar, 3.4 mm. “Reprinted from Neuron, Vol 12, number 3, H. Monyer, N Burnashev, D.J. Laurie, B. Sakmann, P.H. Seeburg, Developmental and regional expression in the rat brain and functional properties of four NMDA receptors, Pages No. 529-524, Copyright (1994), with permission from Elsevier”.
2. NMDA Receptors in Schizophrenia
2.1. Clinical Evidence
The first clue of the implication of NMDARs in schizophrenia resulted from the observations that NMDAR blockers, such as phencyclidine (PCP) and ketamine, induced in healthy individuals psychotic and negative symptoms, as well as cognitive impairment, that resemble those present in schizophrenia [
20,
21,
22,
23] and exacerbated these symptoms in schizophrenic patients [
24,
25]. In addition, ketamine also induced a reduction of NMDA receptors in the human brain, which strongly correlated with negative symptoms [
26]. More recently, neuroimaging studies have shown, for the first time, direct in vivo evidence of a reduction of NMDA receptors in the left hippocampus of medication-free schizophrenic patients [
27]. Schizophrenic patients also exhibit deficits in the attention and information (cognitive and sensorial) processing measured through the prepulse inhibition (PPI) of the acoustic startle response [
28,
29,
30]. PPI is known to prevent the organism from receiving an overload of information, a behavioral event altered in schizophrenia, which is reflective of abnormal functioning of the corticostriato-thalamocortical circuitry. Therefore, thalamic gating deficits would result in an excessive transfer of information to cortical structures and the subsequent cognitive deficiency. However, reductions in PPI were not observed in healthy individuals after the administration of ketamine [
31], which is in sharp contrast to the effects consistently seen in rodents (see below). Another electrophysiological operation impaired in schizophrenia is mismatch negativity (MMN). MMN is an auditory event-related response in an electroencephalographic (EEG) signal, which occurs when a sequence of repetitive sounds is interrupted by an occasional “oddball” sound that differs in frequency (pitch) or duration. This sensory (auditory) information processing is also damaged, not only in schizophrenic patients [
32], but also in their relatives [
33,
34], and it is reported to represent error in prediction. Interestingly, healthy individuals exhibit MMN after a single dose of ketamine [
35]. Taking all these findings together, it is evident that not all components of the symptoms of schizophrenia in human beings are caused by a direct hypofunction of NMDARs. Hence, it appears that MMN is dependent on NMDAR blockade, whereas PPI is not. Further research is needed to understand why these actions are species specific.
2.1.1. Neurophysiology
Brain oscillations have been also studied as possible in vivo biomarkers of the illness. Schizophrenic patients frequently show EEG abnormalities [
36,
37] and a closer look of these changes can bring some evidence about the pathogenesis of the illness. Oscillations in the γ band (30–100 Hz) have been the object of interest, because of their involvement in cognitive functions known to be impaired in schizophrenia [
37,
38,
39,
40,
41,
42,
43]. Cortical γ oscillations result from the control that parvalbumin-containing γ-aminobutyric acid (GABA)ergic interneurons exert over pyramidal neurons [
44,
45] and, in this regard, abnormalities in these cortical interneurons have been consistently found in schizophrenia [
42,
46,
47,
48]. A preferential blockade of NMDA receptors on parvalbumin-expressing (PV) interneurons is postulated as a central mechanism of classical actions of NMDA receptor antagonists, which results in increased cortical activity and γ-band oscillations [
49,
50,
51,
52,
53]. Further, schizophrenia is characterized by abnormalities in γ oscillations measured in different cognitive tasks. Thus, schizophrenic patients exhibit increases in spontaneous gamma band power [
42,
43], which have been related to positive symptoms [
54,
55,
56]. However, marked deficits in the γ frequency band were observed when γ oscillations were examined under different tasks (for instance, auditory-evoked responses) [
39,
42,
57,
58]. However, at variance to what happen in rodents (see below), NMDAR antagonists, such as ketamine, usually elicit increases in spontaneous γ band power, probably evoked by an excessive stimulation of glutamatergic transmission in cortical and subcortical areas [
42,
43,
59,
60,
61]. Although no direct measure of brain glutamate level can be determined from the brain of schizophrenics, some indirect estimates have implicated increased activation of prefrontal cortex in first-episode schizophrenia [
62,
63,
64] and after ketamine administration [
65,
66], which can be taken as suggestive of increased glutamate release in the human brain.
In addition to high frequency oscillations, low frequency oscillations (α, θ and δ bands) are also related to cognitive processing. For instance, the α wave range (8–12 Hz) is related to working memory processes [
67], whereas the θ range (5–8 Hz) is involved in attention and signal detection [
68], which reflects cortico-hippocampal interactions [
69,
70,
71]. On the other hand, oscillations in the δ band are also involved in decision making procedures [
68].
2.1.2. Post-Mortem Studies
Another important line of investigation aimed at examining the implication of NMDAR in schizophrenia has been the study of post-mortem tissue. Thus, decreased cortical expression of NMDAR subunits have been observed in subjects with schizophrenia, though not in a consistent manner [
72], depending on the brain region examined and methodology used. In this regard, decreased transcripts coding for the GluN1 subunit have been found in the prefrontal cortex [
73,
74] and hippocampal subregions [
75,
76]. However, as aforementioned, care must be taken, because some of these changes are also observed in different psychiatric conditions and, by no means, are compelling, as long as contradictory results have been found. As a matter of fact, this same meta-analysis found no consistent statistically significant changes in cortical mRNA and protein expression of GluN2A, GluN2B and GluN2D subunits in schizophrenia, with the exception of decreased expression of mRNA coding for GluN2C [
74,
77,
78]. Of note, in the postsynaptic density compartment of human post-mortem prefrontal cortex, an important reduction in the density of the postsynaptic protein PSD-95 [
74] and in the activity of signaling cascades downstream of the NMDAR has been found in schizophrenia [
79].
Since the finding that the therapeutic efficacy of antipsychotic drugs was directly correlated to their affinity for dopamine D2 receptors [
80,
81], it was first postulated that altered dopamine D2-like receptors was responsible for schizophrenia symptoms. However, this was not confirmed until the study by Abi-Dargham [
82], which reported increased occupancy of dopamine D2 receptors in schizophrenia. Moreover, it was evidenced that the administration of ketamine to healthy subjects enhanced the release of dopamine in ventral striatum, which was shown to correlate strongly with the emergence of psychotic symptoms [
83]. For these reasons, an association between D2-like receptors and NMDA hypofunction was hypothesized [
84,
85]. In addition to these findings, the differential expression of some splice variants might also evoke abnormal NMDAR trafficking and plasma membrane insertion, which may lead to abnormalities seen in schizophrenia patients [
86,
87].
Another important issue is the role of astrocytes in schizophrenia. High levels of extracellular glutamate are associated with excitotoxicity, and astrocytes are the principal cells that contribute to glutamate homeostasis through its removal from the synapsis, by means of selective reuptake mechanisms. The excitatory amino acid transporters 1 and 2 (EAAT1/2) are predominantly localized on astrocytes and are mainly responsible for clearing synaptic glutamate and influencing postsynaptic responses. Post-mortem studies have reported that EAAT1 expression is decreased in schizophrenia, as compared to healthy subjects [
88]. Additionally, the activation of EAAT2 and its transport to plasma membrane was found to be reduced in schizophrenic brains [
89]. A genetic variant of EAAT2 (SNP rs4354668) has been correlated with the severity of schizophrenia [
90]. Altogether, these findings are consistent with an impairment of astrocyte function in schizophrenia [
91].
2.2. Preclinical Evidence
Mental disorders, such as schizophrenia and depression, are illnesses uniquely human, in that they are diagnosed using different interview questionnaires. Therefore, no animal model can recreate the full spectrum of their symptoms. However, some models exist that examine changes in behavioral and neurophysiological readouts that are compatible with changes that are also observed in patients. For instance, the acute, systemic administration of NMDAR antagonists induces, in rodents, hyperlocomotion and stereotypical behaviors [
92], which are potentially compatible to positive symptoms of schizophrenia [
93,
94], in that they are associated with increased dopaminergic and serotonergic transmission in the brain [
95,
96]. NMDAR antagonists, such as PCP and dizocilpine (MK-801), also produce severe disruptions in PPI, and deficits in different domains of cognition in rats [
97,
98]. Acute NMDAR antagonism also evokes an increased firing rate of pyramidal neurons and expression of c-fos mRNA of the prefrontal cortex [
99,
100,
101,
102,
103], which suggests an overall, excessive prefrontal activity, which results in elevated release of glutamate [
104,
105,
106], dopamine [
107,
108,
109], 5-HT [
110,
111,
112] and acetylcholine [
113,
114] in the medial prefrontal cortex (mPFC) of rats. Altogether these findings indicate that an overstimulation of different transmitter systems in the mPFC is a general response to NMDAR hypofunction, which can account for the behavioral effects induced by NMDAR antagonists. The enhanced release of monoamines most likely result from the stimulation of prefrontal excitatory glutamatergic inputs onto midbrain dopamine and 5-HT cell groups, as suggested by recent investigations [
115,
116]. It is paradoxical that a blockade of excitatory glutamatergic receptors, such as NMDARs, results in the stimulation of glutamate release. The most accepted hypothesis to explain this phenomenon is the disinhibition theory [
49,
104,
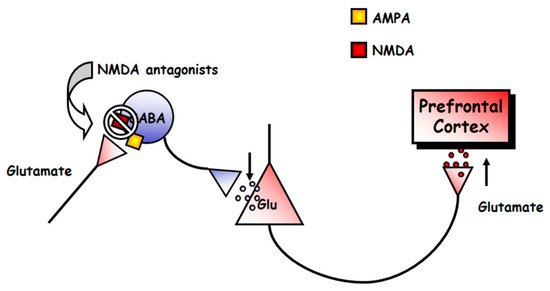
117], which postulates that NMDA antagonists would block NMDA receptors located on tonically active GABAergic neurons, which would control glutamatergic output. This would diminish GABAergic inhibition, thus disinhibiting glutamatergic neurotransmission impinging upon α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (). The finding that some GABAergic interneurons in hippocampus and neocortex are enriched in NMDAR and receive a greater glutamatergic input in comparison with pyramidal neurons, gives further cellular support to this view [
118,
119].
Figure 3. Scheme of the mechanism of action of NMDAR antagonists. These drugs would block NMDAR in a population of tonically active γ-aminobutyric acid (GABA)ergic neurons. This would decrease the activity of these neurons, and the consequent decrease in GABA release would cause the disinhibition of glutamatergic neurotransmission. The glutamate released would lead to the stimulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in pyramidal cells, which would result in the described state of hyperactivity.
Interestingly, it has been reported that an action of NMDAR antagonists on the mPFC of both brain hemispheres is needed to model these changes in the mPFC [
102]. In addition, transmitter changes have been found in other brain areas. For instance, noncompetitive NMDAR antagonists also enhance the efflux of glutamate in the nucleus accumbens [
120], acetylcholine in the retrosplenial cortex and hippocampus [
121,
122], 5-HT in the nucleus accumbens [
111], noradrenaline in nucleus accumbens and hippocampus [
123,
124,
125] and dopamine in limbic areas, such as the nucleus accumbens, hippocampus and ventral pallidum [
108,
111,
113,
126,
127,
128], although these effects were less pronounced than in the mPFC.
2.2.1. Neurophysiology
Abnormal oscillatory patterns have been also observed in rodent models. Hence, systemic administration, and even the local administration of PCP, MK-801 or ketamine into some brain regions, increase γ and high frequency oscillations (HFO) in a number of cortical and subcortical structures (see [
129], for a review). It has also been observed that ketamine and MK-801 reduced the frequency and power of θ oscillations in the hippocampus [
130,
131]. Furthermore, acute PCP also alters thalamo-cortical oscillations, particularly those below the 4 Hz band [
132].
2.2.2. Animal Models
The modeling of schizophrenia has been achieved not only by acute, but also by long-term administration of NMDAR antagonists [
133]. Thus, although the nature of some changes is similar, following acute or protracted treatment with NMDAR antagonists, it has been postulated that changes after the acute regimen are more comparable with those occurring in early stages of schizophrenia, whereas the duration of such changes after sustained administration appears to be more related to the persistence of clinical symptoms of the illness [
134,
135,
136,
137,
138]. Acute PCP treatment increased locomotor activity in rodents, an effect potentiated after long-term treatment [
139,
140,
141]. Subchronic PCP treatment does not seem to affect basal and PCP-induced 5-HT efflux in the mPFC. However, in comparison to acute administration, subchronic PCP attenuated basal prefrontal dopamine release, but potentiated PCP-induced dopamine efflux. The reduced basal extracellular concentration of dopamine could be accounted for by lowering its synthesis, as measured by a diminished expression of tyrosine hydroxylase mRNA in the ventral tegmental area [
141].
Preclinical evidence from animal models also reported impairment of astrocyte function in the schizophrenia model of repeated MK-801 exposure [
142]. In addition, abnormal EAAT1/2 function is associated with schizophrenia phenotypes. For instance, mice lacking EAAT1 showed hyperlocomotion and increased sensitivity to the locomotor hyperactivity produced by NMDAR antagonists [
143]. In addition, the hyperlocomotion of these EAAT1 knockout mice was reversed by the antipsychotic haloperidol.
3. NMDA Receptors in Depression
3.1. Clinical Evidence
Four different findings suggest a relationship between dysfunction of NMDAR and depression. First, an anomalous gene expression of NMDAR has been found in depressed people [
165,
166,
167]; second, stressors induce excessive NMDAR activity that could result in the pathology of depression [
168]; third, NMDAR blockers, such as ketamine (see below), have antidepressant properties [
169,
170,
171]; and fourth, conventional antidepressant drugs usually impact on NMDAR function [
172,
173,
174]. Altogether, these investigations suggest an overstimulation of NMDAR in major depression [
175,
176,
177,
178].
Post-Mortem Studies
However, the results obtained from these investigations exhibit great variability, and are far from consistent, likely due to differences in the brain structure examined or the methodological procedures used. For instance, post-mortem studies have revealed reduced levels of GluN2A and GluN2B subunits in the prefrontal [
179] and perirhinal [
180] cortices, but increased levels of GluN2A subunits in the lateral amygdala [
181] in major depression. Previous post-mortem work did not find changes in the total content of GluN1 protein in the prefrontal cortex in depression [
179,
182]. Yet, when splice isoforms were considered, NMDAR activity and the GluN1 subunit carrying the C1 cytosolic segment were found to be increased in depressives [
182]. Another report also described that the GluN2C subunit is elevated in the locus coeruleus of patients with major depression [
183]. Further, early life adverse effects reduce NMDAR binding in dorsolateral prefrontal and anterior cingulate cortices, which could result from excessive NMDAR stimulation [
184]. This would be consistent with the hypothesis of glutamate excitotoxicity produced by stress-induced excessive NMDAR activity, which could induce depressive states [
168,
185].
Further work has reported higher expression levels of the NMDAR subunit genes, GRIN2B and GRIN2C, in the locus coeruleus of depressed patients [
186]. Another study reported a higher expression of GRIN1, GRIN2A, GRIN2B, GRIN2C and GRIN2D subunit mRNAs, but only in female MDD patients. Nevertheless, when male and female patients were grouped, the expression of GRIN2B mRNA was higher in those who committed suicide, in comparison with those that suffered depression but did not die by suicide [
187]. For this reason, GRIN2B mRNA level is rather considered as biomarker of suicide and, in fact, polymorphisms of GRIN2B have been postulated to predict treatment-resistant depression [
165,
188]. Further epigenetic work showed that methylation in GRIN1 was a significant predictor of depression in a sample of maltreated children [
189].
Although there is a great number of genome wide association studies (GWAS) that have examined genetic changes in depression, much less attention so far has been devoted to the study of gene methylation. In this regard, only one study has found a hypermethylation of the GRIN2A gene body in the hippocampus and prefrontal cortex of post-mortem human tissue in depression [
167], which has been attributed to overexpression of the GluN2A subunit [
190].
Other post-mortem studies have revealed the influence of astrocytes in MDD (see [
191,
192] for review). Thus, there is mounting evidence that the number and morphology of astrocytes are altered in depression, particularly in the frontal cortex [
193,
194,
195] and the dentate gyrus of the hippocampus [
196]. A significant reduction of the packing density of glial fibrillary acidic protein (GFAP)-containing astrocytes was also found, but only in younger (30–45 years old) patients [
197].
3.2. Preclinical Evidence
There is substantial evidence from rodent models relevant to depression that stress induces glutamatergic hyperactivity, as well as the overexpression of NMDARs [
198,
199,
200]. Given that NMDAR antagonists exert a preferential blockade of NMDAR on PV interneurons, enhanced PV interneuron activity has been observed after stress and might underlie depression-like behavior [
201,
202,
203]. However, this is not a universal picture inasmuch as decreased PV cell activity has been observed after different stressful conditions (see [
204], for review).
Maternal separation induces increased expression of GluN2A (but not GluN2B) subunit of NMDARs in the hippocampus of adult rats [
205]. Chronic restraint stress significantly elevated GRIN2a (GluN2A) and GRIN2b (GluN2B) subunit genes in BALB/c mice, but not in C57BL/6 mice [
206]. Chronic restraint stress also increased the levels of GRIN1 mRNA, along with a reduction in protein levels in dorsal hippocampus [
207]. Chronic corticosterone administration, which emulates the endocrine response to stress, increased GRIN2A and GRIN2B mRNAs, which mediated the deleterious effects on the hippocampus [
208]. Further, the olfactory bulbectomy model of depression reduces NMDA receptor binding in the prefrontal cortex and amygdala [
209,
210]. On the other hand, in the frontal cortex, BDNF deficiency, which occurs under chronic stress and is one of the leading causes of depression, also increased the density of GRIN1, GRIN2A and GRIN2B genes in the early stages of development [
211]. Therefore, with all these findings taken together, a logical reasoning would hypothesize that the deletion or inhibition of NMDAR subunits would have antidepressant-like effects. Indeed, inactivation of the GluN2A subunit has been shown to evoke antidepressant-like activity in mice [
212]. Yet, the deletion of the GluN2D subunit in the bed nucleus of the stria terminalis (BNST) increases depressive-like behaviors [
213]. However, mice with constitutive, global deletion of the GluN1 or GluN2B subunits die neonatally. Homozygous GRIN1 knockout mice only survive 8–15 h after birth [
214], and homozygous GRIN2b knockout mice die at early postnatal stages, because of an impaired suckling response [
215].
However, the same as occurs with human studies, work with experimental animals has also yielded contradictory results. Thus, increased GRIN1 mRNA expression in the mPFC appears to have antidepressant-like effects in the forced swim test. [
216]. GRIN1 was also found to be downregulated in chronic unpredictable mild stress (CUMS) [
217]. The type and duration of the stressor, as well as the brain region examined, may underlie these differences.
Alterations in the number and/or function of astrocytes have also been found in animal models of depression (see [
218] for review). Indeed, a reduced number and volume of astrocytes was found in the prefrontal cortex [
219] and the hippocampus [
220], in the chronic unpredictable stress procedure and chronic social defeat stress paradigm. Interestingly, neurotoxic lesioning of astrocytes in the prefrontal cortex is sufficient to induce depressive-like behaviors in rodents [
221], an effect also found after knocking-down the expression of astrocytic glutamate transporter GLAST/GLT-1 in the prefrontal cortex of the mouse, using small interfering RNA (siRNA) strategies [
222].
This entry is adapted from the peer-reviewed paper 10.3390/biom10060947