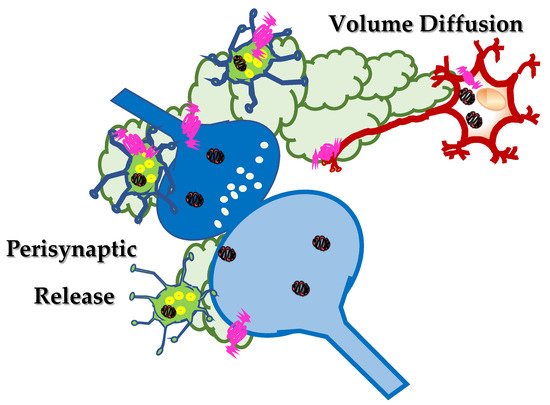
Somatostatin is widely diffused in the central nervous system, where it participates to control the efficiency of synaptic transmission. This peptide mainly colocalizes with GABA, in inhibitory, GABA-containing interneurons from which it is actively released in a Ca2+ dependent manner upon application of depolarizing stimuli. Once released in the synaptic cleft, somatostatin acts locally, or it diffuses in the extracellular space through “volume diffusion”, a mechanism(s) of distribution which mainly operates in the cerebrospinal fluid and that assures the progression of neuronal signalling from signal-secreting sender structures towards receptor-expressing targeted neurons located extrasynaptically, in a non-synaptic, inter-neuronal form of communication. Somatostatin controls the efficiency of central glutamate transmission by either modulating presynaptically the glutamate exocytosis or by metamodulating the activity of glutamate receptors colocalized and functionally coupled with somatostatin receptors in selected subpopulations of nerve terminals. Deciphering the role of somatostatin in the mechanisms of “volume diffusion” and in the “receptor-receptor interaction” unveils new perspectives in the central role of this fine tuner of synaptic strength, paving the road to new therapeutic approaches for the cure of central disorders.
- somatostatin
- glutamate
- noradrenaline
- metamodulation
- volume diffusion
- NMDA receptors
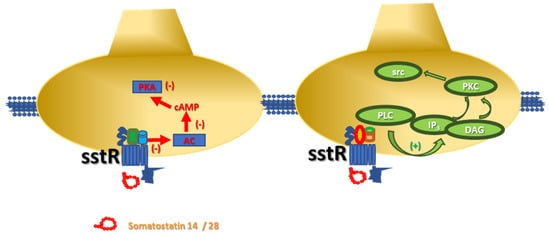
- sst2 receptors
- sst5 receptors
1. The Somatostatinergic System in the Central Nervous System
2. Somatostatin in the Central Nervous System: Synaptic Release and Volume Diffusion
3. Somatostatin Receptors and Metamodulation in the Central Nervous System
This entry is adapted from the peer-reviewed paper 10.3390/ijms22115864
References
- Brazeau, P.; Vale, W.; Burgus, R.; Ling, N.; Butcher, M.; Rivier, J.; Guillemin, R. Hypothalamic Polypeptide That Inhibits the Secretion of Immunoreactive Pituitary Growth Hormone. Science 1973, 179, 77–79.
- Reichlin, S. Somatostatin. N. Engl. J. Med. 1983, 309, 1495–1501.
- Zingg, H.H.; Patel, Y.C. Processing of synthetic somatostatin-28 and a related endogenous rat hypothalamic somatostatin-like molecule by hypothalamic enzymes. Life Sci. 1982, 30, 525–533.
- Robbins, R.J.; Reichlin, S. Somatostatin Biosynthesis by Cerebral Cortical Cells in Monolayer Culture. Endocrinology 1983, 113, 574–581.
- Vezzani, A.; Hoyer, D. Brain somatostatin: A candidate inhibitory role in seizures and epileptogenesis. Eur. J. Neurosci. 1999, 11, 3767–3776.
- Günther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.-J.; Lupp, A.; Korbonits, M.; Castaño, J.P.; Wester, H.-J.; et al. International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature. Pharmacol. Rev. 2018, 70, 763–835.
- Gatto, F.; Barbieri, F.; Arvigo, M.; Thellung, S.; Amarù, J.; Albertelli, M.; Ferone, D.; Florio, T. Biological and Biochemical Basis of the Differential Efficacy of First and Second Generation Somatostatin Receptor Ligands in Neuroendocrine Neoplasms. Int. J. Mol. Sci. 2019, 20, 3940.
- Bruns, C.; Weckbecker, G.; Raulf, F.; Lübbert, H.; Hoyer, D. Characterization of Somatostatin Receptor Subtypes. Novartis Found. Symp. 2007, 190, 89–110.
- Siehler, S.; Nunn, C.; Hannon, J.; Feuerbach, D.; Hoyer, D. Pharmacological profile of somatostatin and cortistatin receptors. Mol. Cell. Endocrinol. 2008, 286, 26–34.
- Kumar, U.; Grant, M. Somatostatin and Somatostatin Receptors. Chem. Biol. Pteridines Folates 2009, 50, 97–120.
- Hannon, J.P.; Nunn, C.; Stolz, B.; Bruns, C.; Weckbecker, G.; Lewis, I.; Troxler, T.; Hurth, K.; Hoyer, D. Drug Design at Peptide Receptors. J. Mol. Neurosci. 2002, 18, 15–28.
- Pittaluga, A.; Feligioni, M.; Longordo, F.; Arvigo, M.; Raiteri, M. Somatostatin-Induced Activation and Up-Regulation of N-Methyl-d-aspartate Receptor Function: Mediation through Calmodulin-Dependent Protein Kinase II, Phospholipase C, Protein Kinase C, and Tyrosine Kinase in Hippocampal Noradrenergic Nerve Endings. J. Pharmacol. Exp. Ther. 2004, 313, 242–249.
- Thermos, K.; Bagnoli, P.; Epelbaum, J.; Hoyer, D. The somatostatin sst1 receptor: An autoreceptor for somatostatin in brain and retina? Pharmacol. Ther. 2006, 110, 455–464.
- Yang, L.; Berk, S.C.; Rohrer, S.P.; Mosley, R.T.; Guo, L.; Underwood, D.J.; Arison, B.H.; Birzin, E.T.; Hayes, E.C.; Mitra, S.W.; et al. Synthesis and biological activities of potent peptidomimetics selective for somatostatin receptor subtype 2. Proc. Natl. Acad. Sci. USA 1998, 95, 10836–10841.
- Rohrer, S.P.; Birzin, E.T.; Mosley, R.T.; Berk, S.C.; Hutchins, S.M.; Shen, D.-M.; Xiong, Y.; Hayes, E.C.; Parmar, R.M.; Foor, F.; et al. Rapid Identification of Subtype-Selective Agonists of the Somatostatin Receptor Through Combinatorial Chemistry. Science 1998, 282, 737–740.
- Binaschi, A.; Bregola, G.; Simonato, M. On the Role of Somatostatin in Seizure Control: Clues from the Hippocampus. Rev. Neurosci. 2003, 14, 285–301.
- Hoyer, D.; Bell, G.; Berelowitz, M.; Epelbaum, J.; Feniuk, W.; Humphrey, P.; O’Carroll, A.-M.; Patel, Y.; Schonbrunn, A.; Taylor, J.; et al. Classification and nomenclature of somatostatin receptors. Trends Pharmacol. Sci. 1995, 16, 86–88.
- Tallent, M.K. Presynaptic Inhibition of Glutamate Release by Neuropeptides: Use-Dependent Synaptic Modification. Chem. Biol. Pteridines Folates 2007, 44, 177–200.
- Martel, G.; Dutar, P.; Epelbaum, J.; Viollet, C.P. Somatostatinergic systems: An update on brain functions in normal and pathological aging. Front. Endocrinol. 2012, 3, 154.
- Saha, S.; Henderson, Z.; Batten, T. Somatostatin immunoreactivity in axon terminals in rat nucleus tractus solitarii arising from central nucleus of amygdala: Coexistence with GABA and postsynaptic expression of sst2A receptor. J. Chem. Neuroanat. 2002, 24, 1–13.
- Jinno, S.; Kosaka, T. Patterns of colocalization of neuronal nitric oxide synthase and somatostatin-like immunoreactivity in the mouse hippocampus: Quantitative analysis with optical disector. Neuroscience 2004, 124, 797–808.
- Chesselet, M.; Graybiel, A. Striatal neurons expressing somatostatin-like immunoreactivity: Evidence for a peptidergic interneuronal system in the cat. Neurosci. 1986, 17, 547–571.
- Forloni, G.; Hohmann, C.; Coyle, J.T. Developmental expression of somatostatin in mouse brain. I. Immunocytochemical studies. Dev. Brain Res. 1990, 53, 6–25.
- Eriksson, L.G. An immunohistochemical study of somatostatin and neurotensin positive neurons in the septal nuclei of the rat brain. Brain Struct. Funct. 1984, 170, 1–10.
- Vincent, S.R.; McIntosh, C.H.S.; Buchan, A.M.J.; Brown, J.C. Central somatostatin systems revealed with monoclonal antibodies. J. Comp. Neurol. 1985, 238, 169–186.
- Johansson, O.; Hökfelt, T.; Elde, R. Immunohistochemical distribution of somatostatin-like immunoreactivity in the central nervous system of the adult rat. Neuroscience 1984, 13, 265-IN2.
- Riedemann, T. Diversity and Function of Somatostatin-Expressing Interneurons in the Cerebral Cortex. Int. J. Mol. Sci. 2019, 20, 2952.
- Iversen, L.L.; Iversen, S.D.; Bloom, F.E.; Douglas, C.L.; Brown, M.R.; Vale, W. Calcium-dependent release of somatostatin and neurotensin from rat brain in vitro. Nat. Cell Biol. 1978, 273, 161–163.
- Bonanno, G.; Parodi, B.; Cafaggi, S.; Raiteri, M. Somatostatin Release from Rat Cerebral Cortex Synaptosomes. J. Neurochem. 1991, 57, 1258–1264.
- Raiteri, M.; Angelini, F.; Levi, G. A simple apparatus for studying the release of neurotransmitters from synaptosomes. Eur. J. Pharmacol. 1974, 25, 411–414.
- Raiteri, L.; Raiteri, M. Synaptosomes Still Viable after 25 Years of Superfusion. Neurochem. Res. 2000, 25, 1265–1274.
- Freund, T.F.; Buzsáki, G. Interneurons of the Hippocampus. Hippocampus 1996, 6, 347–470.
- Mathé, A.A.; Nomikos, G.G.; Svensson, T.H. In vivo release of somatostatin from rat hippocampus and striatum. Neurosci. Lett. 1993, 149, 201–204.
- Bonanno, G.; Gemignani, A.; Schmid, G.; Severi, P.; Cavazzani, P.; Raiteri, M. Human brain somatostatin release from isolated cortical nerve endings and its modulation through GABAB receptors. Br. J. Pharmacol. 1996, 118, 1441–1446.
- Bonanno, G.; Raiteri, M. Multiple GABAB receptors. Trends Pharmacol. Sci. 1993, 14, 259–261.
- Dournaud, P.; Boudin, H.; Schonbrunn, A.; Tannenbaum, G.S.; Beaudet, A. Interrelationships between Somatostatin sst2A Receptors and Somatostatin-Containing Axons in Rat Brain: Evidence for Regulation of Cell Surface Receptors by Endogenous Somatostatin. J. Neurosci. 1998, 18, 1056–1071.
- Dutar, P.; Vaillend, C.; Viollet, C.; Billard, J.-M.; Potier, B.; Carlo, A.-S.; Ungerer, A.; Epelbaum, J. Spatial learning and synaptic hippocampal plasticity in type 2 somatostatin receptor knock-out mice. Neuroscience 2002, 112, 455–466.
- Mastrodimou, N.; Thermos, K. The somatostatin receptor (sst1) modulates the release of somatostatin in rat retina. Neurosci. Lett. 2004, 356, 13–16.
- Agnati, L.F.; Cortelli, P.; Biagini, G.; Bjelke, B.; Fuxe, K. Different classes of volume transmission signals exist in the central nervous system, and are affected by metabolic signals, temperature gradients, and pressure waves. NeuroReport 1994, 6, 9–12.
- Vizi, E.; Kiss, J.P.; Lendvai, B. Nonsynaptic communication in the central nervous system. Neurochem. Int. 2004, 45, 443–451.
- Herkenham, M. Mismatches between neurotransmitter and receptor localizations in brain: Observations and implications. Neuroscience 1987, 23, 1–38.
- Sun, Q.-Q.; Huguenard, J.R.; Prince, D.A. Somatostatin Inhibits Thalamic Network Oscillations In Vitro: Actions on the GABAergic Neurons of the Reticular Nucleus. J. Neurosci. 2002, 22, 5374–5386.
- Chen, W.; Ke, J.-B.; Wu, H.-J.; Miao, Y.; Li, F.; Yang, X.-L.; Wang, Z. Somatostatin receptor-mediated suppression of gabaergic synaptic transmission in cultured rat retinal amacrine cells. Neuroscience 2014, 273, 118–127.
- Momiyama, T.; Zaborszky, L. Somatostatin Presynaptically Inhibits Both GABA and Glutamate Release Onto Rat Basal Forebrain Cholinergic Neurons. J. Neurophysiol. 2006, 96, 686–694.
- Fuxe, K.; Agnati, L.F. Receptor-receptor interactions in the central nervous system. A new integrative mechanism in synapses. Med. Res. Rev. 1985, 5, 441–482.
- Borroto-Escuela, D.O.; Tarakanov, A.O.; Brito, I.; Fuxe, K. Glutamate heteroreceptor complexes in the brain. Pharmacol. Rep. 2018, 70, 936–950.
- Fuxe, K.; Borroto-Escuela, D.O. Volume transmission and receptor-receptor interactions in heteroreceptor complexes: Understanding the role of new concepts for brain communication. Neural Regen. Res. 2016, 11, 1220–1223.
- Fuxe, K.; Borroto-Escuela, D.O.; Romero-Fernandez, W.; Palkovits, M.; Tarakanov, A.O.; Ciruela, F.; Agnati, L.F. Moonlighting Proteins and Protein–Protein Interactions as Neurotherapeutic Targets in the G Protein-Coupled Receptor Field. Neuropsychopharmacology 2013, 39, 131–155.
- Ribeiro, J.A.; Sebastião, A.M. Modulation and metamodulation of synapses by adenosine. Acta Physiol. 2010, 199, 161–169.
- Olivero, G.; Vergassola, M.; Cisani, F.; Roggeri, A.; Pittaluga, A. Presynaptic Release-regulating Metabotropic Glutamate Receptors: An Update. Curr. Neuropharmacol. 2020, 18, 655–672.
- Di Prisco, S.; Olivero, G.; Merega, E.; Bonfiglio, T.; Marchi, M.; Pittaluga, A. CXCR4 and NMDA Receptors Are Functionally Coupled in Rat Hippocampal Noradrenergic and Glutamatergic Nerve Endings. J. Neuroimmune Pharmacol. 2016, 11, 645–656.
- Olivero, G.; Vergassola, M.; Cisani, F.; Usai, C.; Pittaluga, A. Immuno-Pharmacological Characterization of Presynaptic GluN3A-Containing NMDA Autoreceptors: Relevance to Anti-NMDA Receptor Autoimmune Diseases. Mol. Neurobiol. 2019, 56, 6142–6155.
- Pittaluga, A. Presynaptic release-regulating NMDA receptors in isolated nerve terminals: A narrative review. Br. J. Pharmacol. 2021, 178, 1001–1017.
- Olivero, G.; Grilli, M.; Vergassola, M.; Bonfiglio, T.; Padolecchia, C.; Garrone, B.; Di Giorgio, F.P.; Tongiani, S.; Usai, C.; Marchi, M.; et al. 5-HT2A-mGlu2/3 receptor complex in rat spinal cord glutamatergic nerve endings: A 5-HT2A to mGlu2/3 signalling to amplify presynaptic mechanism of auto-control of glutamate exocytosis. Neuropharmacology 2018, 133, 429–439.
- Durán-Prado, M.; Malagón, M.M.; Gracía-Navarro, F.; Castaño, J.P. Dimerization of G protein-coupled receptors: New avenues for somatostatin receptor signalling, control and functioning. Mol. Cell. Endocrinol. 2008, 286, 63–68.
- Blake, A.C.B.A.M.Z.S.A.D.; Badway, A.; Strowsk, M. Delineating Somatostatins Neuronal Actions. Curr. Drug Target -CNS Neurol. Disord. 2004, 3, 153–160.
- Moneta, D.; Richichi, C.; Aliprandi, M.; Dournaud, P.; Dutar, P.; Billard, J.M.; Carlo, A.S.; Viollet, C.; Hannon, J.P.; Fehlmann, D.; et al. Somatostatin receptor subtypes 2 and 4 affect seizure susceptibility and hippocampal excitatory neurotransmission in mice. Eur. J. Neurosci. 2002, 16, 843–849.
- Gastambide, F.; Lepousez, G.; Viollet, C.; Loudes, C.; Epelbaum, J.; Guillou, J.-L. Cooperation between hippocampal somatostatin receptor subtypes 4 and 2: Functional relevance in interactive memory systems. Hippocampus 2009, 20, 745–757.
- Cammalleri, M.; Liu, Y.; Monte, M.D.; Cervia, D.; Langenegger, D.; Hoyer, D.; Bagnoli, P. Somatostatin receptors differentially affect spontaneous epileptiform activity in mouse hippocampal slices. Eur. J. Neurosci. 2004, 20, 2711–2721.
- Cammalleri, M.; Martini, D.; Timperio, A.M.; Bagnoli, P. Functional effects of somatostatin receptor 1 activation on synaptic transmission in the mouse hippocampus. J. Neurochem. 2009, 111, 1466–1477.
- Pfeiffer, M.; Koch, T.; Schröder, H.; Laugsch, M.; Höllt, V.; Schulz, S. Heterodimerization of Somatostatin and Opioid Receptors Cross-modulates Phosphorylation, Internalization, and Desensitization. J. Biol. Chem. 2002, 277, 19762–19772.
- Baragli, A.; Alturaihi, H.; Watt, H.L.; Abdallah, A.; Kumar, U. Heterooligomerization of human dopamine receptor 2 and somatostatin receptor 2: Co-immunoprecipitation and fluorescence resonance energy transfer analysis. Cell. Signal. 2007, 19, 2304–2316.
- Rocheville, M.; Lange, D.C.; Kumar, U.; Patel, S.C.; Patel, R.C.; Patel, Y.C. Receptors for Dopamine and Somatostatin: Formation of Hetero-Oligomers with Enhanced Functional Activity. Science 2000, 288, 154–157.