Proliferating cell nuclear antigen (PCNA) is an essential factor in DNA replication and repair. It forms a homotrimeric ring that embraces the DNA and slides along it, anchoring DNA polymerases and other DNA editing enzymes. It also interacts with regulatory proteins through a sequence motif known as PCNA Interacting Protein box (PIP-box). We here review the latest contributions to knowledge regarding the structure-function relationships in human PCNA, particularly the mechanism of sliding, and of the molecular recognition of canonical and non-canonical PIP motifs. The unique binding mode of the oncogene p15 is described in detail, and the implications of the recently discovered structure of PCNA bound to polymerase δ are discussed. The study of the post-translational modifications of PCNA and its partners may yield therapeutic opportunities in cancer treatment, in addition to illuminating the way PCNA coordinates the dynamic exchange of its many partners in DNA replication and repair.
1. DNA Sliding Clamps
Replicative DNA polymerases are the enzymes that replicate chromosomal DNA during the S-phase of the cell cycle. They can quickly polymerize thousands of nucleotides without detaching from the genomic template [
1]. This fast and processive activity is conferred to polymerases by their association with multimeric ring shaped proteins known as DNA sliding clamps. They encircle and slide along the DNA, tethering polymerases and other factors to the DNA duplex [
2].The first evidence of the sliding clamp structure was obtained from the polymerase III β subunit of the
E. coli Pol-III complex. Biochemical assays demonstrated that the β subunit interacts tightly with nicked circular plasmid, while it dissociates when the DNA is linearized [
3]. DNA Sliding clamps are loaded onto DNA by clamp loaders, evolutionarily conserved proteins of the AAA+ family of ATPases [
4,
5]. They form pentameric complexes that bind and open the sliding clamp, placing it onto the DNA 3′ end of the primer/template-junction, in an ATP dependent process [
6].
Sliding clamps are functionally and structurally similar across all living organisms, including some viruses, and can assemble in homodimeric, homotrimeric, or heterotrimeric rings, with the protomers binding each other in a head-to-tail fashion [
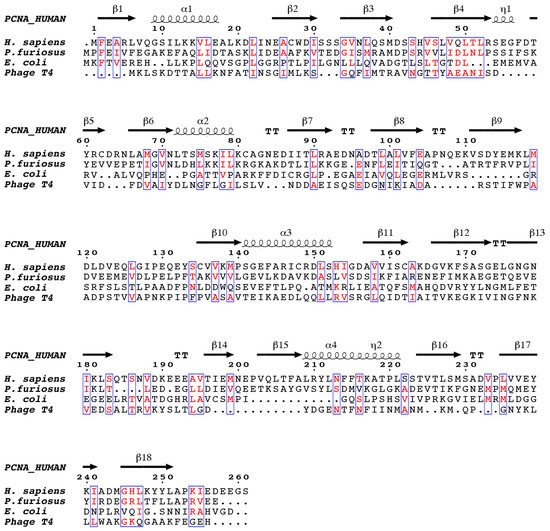
7]. Despite the low sequence similarity between the sliding clamps of different domains of life (), they all adopt a three-dimensional pseudo-six fold symmetry structure consisting of an outer layer of 6 β-sheets and an inner layer of 12 α-helices facing the central channel [
8,
9].
Figure 1. The structure-based sequence alignment of DNA sliding clamps from H. sapiens, P. furiosus, E. coli, and the gp45 gene of bacteriophage T4. The alignment was performed with chain A from each of the PDB files, which corresponds to one of the three protomers (in the case of E. coli, which consists of two protomers, only the N-terminal two thirds of the sequence are shown). Similar residues are colored red. Secondary structure elements corresponding to human proliferating cell nuclear antigen (PCNA) are shown above the alignment, β-strands are indicated as arrows, α-helices as spirals, and β-turns as TT. The figure was generated with ESPript.
The bacterial β clamp is a homodimeric ring that comprises two protomers [
10], each one with three topologically similar domains. By contrast, the functional equivalents of β clamp in T4 bacteriophage (gene 45 protein, gp45) and PCNA (Proliferating Cell Nuclear Antigen) in eukaryotes and archaea assemble in trimeric rings, each protomer containing two similar domains connected by an interdomain-connecting loop (IDCL) [
11,
12] ().
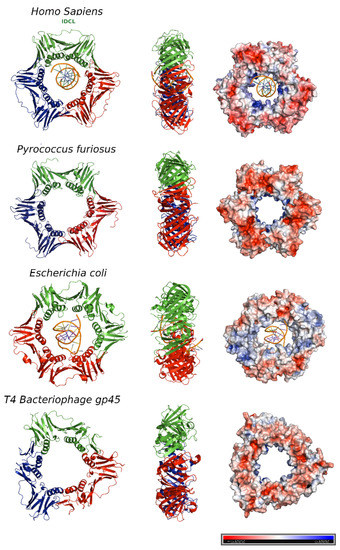
Figure 2. DNA sliding clamps from different organisms. The crystal structures of Homo sapiens PCNA (Protein Data Bank [PDB] code: 6GIS) bound to DNA, Pyrococcus furiosus PCNA (PDB code:1GE8), the Escherichia coli β clamp (PDB code: 3BEP) bound to DNA, and the gene 45 antigen of Bacteriophage T4 (PDB code: 1CZD). For each organism, the front (left) and side views (middle) of the three-dimensional crystal structure are shown. Each protomer is represented by a different color (blue, red and green). The surface electrostatic potential is represented, with positive potential depicted in blue and negative potential in red (right). The potential varies from −5KBT/e to +5KBT/e.
In the T4 bacteriophage ring, the two domains are less similar to each other, and consequently, the gp45 clamp has a triangular appearance instead of the hexagonal shape of the others.
The central pore has an internal diameter of approximately 35 Å, larger than the 24 Å of the double stranded DNA helix (dsDNA) in the canonical B-form [
13]. Overall, clamps are acidic proteins with net negative charges. The outer surface possesses a negative electrostatic potential, but the α-helices facing the central cavity are rich in positively charged amino acids and generate a positive electrostatic potential that allows the DNA to pass through [
14] (, right panel). The negatively charged external surface might contribute to preventing non-specific interactions, facilitating the correct disposition of the DNA inside the ring.
For several years, considerable efforts have been made to structurally assess the association between sliding clamps and DNA, as well as to understand the molecular mechanism by which these ring-shape, multimeric proteins slide along DNA. However, the weakness and lack of sequence specificity of PCNA-DNA interactions made this task difficult. The first available high resolution crystallographic structure of a sliding clamp in complex with DNA was of the
E. coli DNA polymerase III β subunit bound to a designed 10 bp dsDNA [
15] (). In order to mimic the primed DNA strand while being copied by the polymerase, the designed dsDNA had a four-base long 5′-overhang of ssDNA on one strand. The dsDNA portion appeared inside the ring, tilted to an angle of 22°, which could be explained by the contact of the front side of the ring with the DNA, but also by the interaction of the ssDNA with another symmetry related ring in the crystal lattice. The authors proposed that the β-clamp-ssDNA contact acts as a “placeholder”, attaching the clamp at the 3′ end of the primed site and preventing it from sliding off the DNA before the initiation of replication [
15].
The crystal structure of a single-chain chimera of
S. cerevisiae PCNA bound to primed DNA was also solved, but little information could be obtained from the DNA structure due to low occupancy and the presence of disordered regions [
16]. Nevertheless, the model showed dsDNA within the central cavity of the PCNA ring, facilitating contact of the negatively charged phosphate backbone of DNA with positively charged residues on the inner side of PCNA. An ssDNA overhang, in this case, was not observed.
2. Human PCNA Structure
Proliferating cell nuclear antigen (PCNA) was concurrently discovered by two different groups. On one hand, Miyachi et al. [
17] detected an auto-antigen in the sera of some patients with systemic lupus erythematosus, and because the protein was detected in the nuclei of dividing cells, they named it PCNA [
18]. On the other hand, Bravo and Celis [
19] identified a protein which was synthesized during the S-phase cell cycle and called it cyclin. Further experiments showed that both were the same protein of 29 kDa, which behaves as a homotrimer in solution. The PCNA 87 kDa ring is opened by the clamp loader Replication Factor C and is placed encircling the DNA duplex in a process where ATP is hydrolyzed [
20]. Extensive studies demonstrate that PCNA is an auxiliary factor for the replication polymerases δ and ε (Pol δ and Pol ε), increasing their processivity by tethering them and sliding along the double-stranded DNA helix. Especially important is its role in the synthesis of the lagging strand, acting as a platform where Pol δ, flap endonuclease 1 (FEN1), and DNA ligase I (LIG1) bind to synthesize, process, and join Okazaki fragments [
21]. PCNA recruits other factors to the replication fork, participating in DNA repair (translesion synthesis, homologous recombination, mismatch repair, and nucleotide excision repair), chromatin remodeling, and cell cycle control [
21]. Not all of its partners bind simultaneously to PCNA, and switching may be triggered by different mechanisms: affinity-competition, proteolysis, or post-translational modifications [
2].
The first crystal structure of human PCNA was solved in 1996, bound to the C-terminal region of the cell-cycle checkpoint protein p21
WAF1/CIP1 (PDB entry: 1AXC) [
22]. However, it was not until 2004 that two structures of the human PCNA trimer alone were elucidated (PDB entries 1VYM and 1W60), corresponding to two distinct spatial groups [
23]. Both structures are essentially the same but a bucking trimer is seen in one, while the other presents a planar ring, indicating a certain degree of plasticity in the spatial arrangement of the PCNA trimer. There are two distinct faces of the ring: the front face, also known as the C-face (since the carboxy-terminal ends of the protomers are located there), contains a hydrophobic pocket next to the IDCL on each protomer, where polymerases and other proteins bind; while the back face has prominent loops emerging into the solvent, and is the target for post-translational modifications that alter the clamp’s properties [
6].
The solution NMR (Nuclear Magnetic Resonance) spectrum of PCNA was assigned [
24], and the backbone chemical shifts and many long range NOEs (Nuclear Overhauser Effect) were measured [
25]. These data were consistent with the crystallographic results, indicating that the structure in solution is the same. Moreover, size exclusion chromatography coupled to multiangle light scattering (SEC-MALS) measurements showed that the trimeric form is predominant even at high concentrations, confirming that recombinant PCNA behaves as one single trimer in solution [
25]. Experiments in intact cells and cell extracts showed that PCNA can form dimers of trimers [
26], but even if PCNA may exist transiently as a loosely bound dimer of trimers inside cells, its functional role is unclear.
Thermal and chemical denaturation studies indicate that although human PCNA has the same three-dimensional structure as the
S. cerevisiae homolog, it is less stable. Human PCNA also displays increased backbone dynamics compared with yeast PCNA, especially at the helices that line the inner surface of the ring. This highly dynamic and plastic behavior of human PCNA could be an evolutionary advantage to facilitate binding to a large number of ligands [
25].
The crystal structure of human PCNA bound to a 10 bp DNA duplex shows the DNA inside the channel, tilted by an angle of 15° relative to the three-fold rotation axis of the ring [
27]. The PCNA-DNA interface comprises six conserved basic residues spread along five α-helices of two protomers that establish polar contacts with five consecutive phosphates of one of the DNA strands. NMR studies confirmed that primed DNA does not bind the hydrophobic pocket of PCNA.
3. PCNA Binding Proteins
A large network of proteins is responsible for replicating DNA with high fidelity and for repairing DNA damage through different pathways. PCNA is a global hub in DNA metabolism that interacts with a large number of proteins involved in a variety of DNA-related processes [
34]. As PCNA is a symmetric homotrimer in solution, it has three identical hydrophobic pockets to simultaneously bind different partners and coordinate a variety of functions in space and time [
35]. Most of the proteins that bind PCNA either are IDPs (intrinsically disordered proteins) or have IDRs (intrinsically disordered regions). IDPs lack defined secondary and tertiary structures under physiological conditions. Although they fulfil important biological functions across all domains of life, they are more abundant in eukaryotes [
36]. In particular, most transcription factors, as well as proteins involved in signal transduction, in eukaryotic organisms are predicted to be disordered or to contain disordered regions [
37]. This reveals a correlation between complex cell regulation and the greater presence of IDPs/IDRs [
38]. Moreover, most of the proteins associated with cancer have been identified as IDP or IDR-containing proteins (79%), which underlines the crucial roles they play in several cellular events that are altered in cancer, such as cell proliferation, DNA repair, and apoptosis [
39]. The evolutionary advantage of IDPs likely lies in their plasticity that allows them to interact with many ligands and their vast regulation through post-translational modifications [
40]. Disordered proteins have large accessible surface areas, which increase their ability to interact with diverse binding partners through short linear motifs (SLiMs) [
41]. Many of the proteins that interact with PCNA display a characteristic SLiM known as PIP box (PCNA interacting protein-box) or an extended version called PIP degron. The canonical PIP box motif is
QXXhXXaa, where
h is an aliphatic hydrophobic residue (frequently I, L, or M) and
a is an aromatic hydrophobic one (F, W, or Y), whereas X can be any amino acid [
9]. The PIP degron motif targets PCNA for degradation and also harbors a basic residue (K or R) four amino acids after the second aromatic residue, as well as a TD motif just before the aromatic residues [
42]. Additionally, a novel motif was identified and found to be present in several PCNA interacting proteins, named KA box, that consists of residues K-A-(A/L/I)-(A/L/Q)-x-x-(L/V) [
43,
44].
Structural studies of PIP box containing proteins or derived peptides in complex with human PCNA have unveiled the molecular details of the PIP motif-PCNA interface. The crystal structure of a p21 fragment bound to human PCNA was the first structural characterization of this interface (PDB code: 1AXC) [
22]. Since then, several co-crystal structures of PCNA with different ligands have been solved. Overall, all PCNA interacting proteins adopt a similar conformation, which consists of an extended N-terminal region, a 3
10 helical turn of four residues enclosed by the hydrophobic residues of the PIP box, and a C-terminal region of variable length that sometimes adopts a β strand secondary structure and interacts with the IDCL. The conserved helix inserts into the hydrophobic pocket of PCNA, whereas the glutamine sticks into the so-called Q-pocket, establishing hydrogen bonds with the backbone of PCNA [
22,
42]. The assignment of the human PCNA NMR spectrum [
24] allowed the analysis of the
1H-
15N correlation spectra of PCNA in the presence of different ligands and the calculation of the perturbations in the NMR signals. Thereby, the NMR data provided complementary or new information regarding these interactions in solution at the residue level [
45].
The available structural information of PCNA in complex with different partners reveals that three ligands are able to bind simultaneously the three identical protomers of the PCNA ring. Isothermal titration calorimetry (ITC) data with different peptides are all well fitted with a model assuming one set of equivalent sites, with no evidence of binding cooperativity. Therefore, it seems that ligands compete for binding with PCNA based on their affinities, which can be modulated by post-translational modifications [
9].
The intrinsically disordered protein p21 is one of the PCNA binding partners showing the highest affinity (), probably because it needs to displace other proteins to block replication in response to DNA damage. This high affinity relies on efficient hydrophobic packing, as well as electrostatic interactions with the C-terminal region of PCNA [
46]. Studies with variable length peptides of p21 suggest that basic residues at the N and C-termini encompassing the PIP box protein also contribute to an increase in the binding affinity. A recent study combining experimental and computational approaches confirmed this correlation between positive patches of residues flanking the PIP box motif and a strong affinity for PCNA [
41]. Moreover, FEN1 endonuclease and p68 (also known as p66, the third subunit of Pol δ) exhibit fewer basic residues than p21, and accordingly present lower affinity (). However, the comparison of the structure of PCNA with full length FEN1 and with a 20-residue long PIP fragment reveals additional contacts involving the core domain and the C-terminal region of FEN1, which increase the affinity of FEN1 by three orders of magnitude.
Table 1. Post-translational modifications of PCNA.
| Modification |
Target Residue |
Enzyme |
Function |
Ref. |
| Monoubiquitination |
K164 |
Rad18, RNF8
CRL4Cdt2 |
Promotes DNA synthesis at damaged sites |
[65] |
| K117 |
Unknown |
Unknown |
[69] |
| Polyubiquitination |
K164 |
Rad5
HLTF/SHPRH |
Promotes TS |
[66] |
| Acetylation |
K13,K14, K77, K80 |
CPB/p300 |
Promotes PCNA degradation after NER |
[31] |
| SUMOylation |
K164 |
UBC9(E2) |
Inhibits DSBs formation |
[68] |
| K254 |
Unknown |
Unknown |
[72] |
| ISGylation |
K164, K168 |
EFP |
Terminates TLS |
[70] |
| NEDDylation |
K164 * |
Rad18 |
Regulates Pol η recruitment
in DDR pathway |
[71] |
| Phosphorylation |
Y114 * |
Unknown |
Promotes adipogenesis in response to fatty diet |
[76] |
| Y211 |
EGR |
Protects PCNA from degradation and inhibits MMR |
[75]
[77] |
| Methylation |
K248 |
SETD8 |
Promotes maturation of Okazaki fragments |
[72] |
| Di-methylation |
K110 * |
EZH2 |
Promotes DNA synthesis by Pol δ |
[73] |
This entry is adapted from the peer-reviewed paper 10.3390/biom10040570