The SARS-CoV-2 pandemic introduced the world to a new type of vaccine based on mRNA encapsulated in lipid nanoparticles (LNPs). Instead of delivering antigenic proteins directly, an mRNA-based vaccine relies on the host’s cells to manufacture protein immunogens which, in turn, are targets for antibody and cytotoxic T cell responses. mRNA-based vaccines have been the subject of research for over three decades as a platform to protect against or treat a variety of cancers, amyloidosis and infectious diseases. In this review, we discuss mRNA-based approaches for the generation of prophylactic and therapeutic vaccines to HIV. We examine the special immunological hurdles for a vaccine to elicit broadly neutralizing antibodies and effective T cell responses to HIV. Lastly, we outline an mRNA-based HIV vaccination strategy based on the immunobiology of broadly neutralizing antibody development.
1. Progress in mRNA Technology for HIV Vaccines
An mRNA vaccine is based on the mRNA encoding of an immunogen(s) that is translated into protein upon delivery into host cells. As early as 1989, it was demonstrated that mRNA encapsulated in cationic liposome was able to transfect mouse cells lines [
106]. Later, it was found that even direct injection of naked mRNA into mouse skeletal muscle results in protein translation and expression [
107,
108]. However, using mRNA for therapeutic or vaccine purposes was not feasible because of the instability of RNA molecules, a lack of efficient delivery methods, uncontrollable activation of innate immunity through RNA sensors and difficulties in large-scale manufacturing of mRNA [
107,
109]. In recent years, technological advances and improved delivery methods have addressed these obstacles and mRNA-based vaccines have emerged as a promising new platform to deliver antigens. Vaccines based on mRNA have several advantages over traditional vaccine platforms, including increased safety, efficacy and ease and speed of manufacture [
8,
10,
110]. In this section, we provide a brief overview of the recent advances in mRNA formulation for vaccines and their exploitation for HIV vaccination.
1.1. Non-Amplifying mRNA Vaccines
Non-amplifying mRNAs, in which the immunogen alone is encoded as an mRNA, represent a simple and economical approach to the development of an mRNA vaccine. Early demonstrations of mRNA-based vaccination relied on non-amplifying mRNAs, injected alone or encapsulated in liposomes, as the source immunogens [
111,
112]. Substantial progress has been made in recent years to increase both the immunogenicity and safety of vaccines based on non-amplifying mRNA, including optimized codon usage, 3′ capping, 3′ and 5′ untranslated regions, poly-A tail, nucleoside modifications and purification method [
8,
113]. Non-amplifying mRNAs are amenable to all of the delivery platforms discussed in
Section 3.2 and have recently received considerable attention as SARS-CoV-2 vaccines when formulated with nucleoside modifications and encapsulated in lipid nanoparticles [
114,
115]. Several pre-clinical and clinical studies of HIV mRNA vaccines have been published () and are discussed in more detail in the context of their delivery method in
Section 3.2.
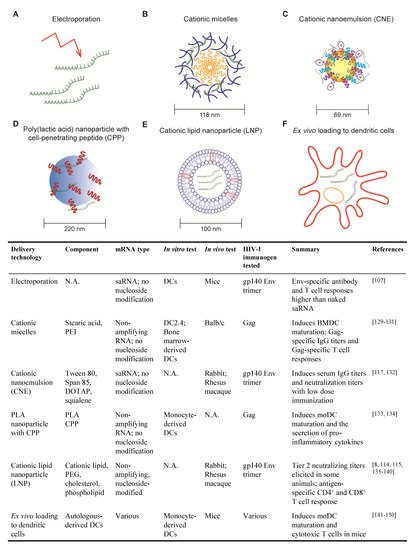
Figure 2. Delivery methods for HIV mRNA vaccine. (A) Electroporation; (B) Cationic micelles composed of stearic acid (yellow) and Polyethylenimine (PEI) (blue); (C) Cationic nanoemulsion (CNE); The yellow core shows squalene. Surfactant such as Tween 80 and Span 85 are shown in blue and purple. Cationic lipid DOTAP (Dioleoyl-3-trimethylammonium propane) is shown with red ‘+’ mark; (D) poly(lactic acid) (PLA) nanoparticle (blue core) with cell penetrating peptide (CPP) (shown in red); (E) Cationic lipid nanoparticle (LNP); figure shows a lipid bilayer with other components that can be included, such as Polyethylene glycol (PEG), cholesterol or phospholipid; (F) Ex vivo loading of dendritic cell (DC). The table below summarizes the HIV mRNA vaccines that have been tested, detailing the delivery method, the type of mRNA, the in vitro and in vivo system used for testing, the mRNA-encoded immunogen and a short note on the results.
1.2. Self-Amplifying RNA Vaccines
Self-amplifying RNAs (saRNAs) are replicons engineered from RNA viruses that encode vaccine immunogens as well as viral replication machinery. As such, saRNAs are capable of replicating their RNAs after entering the cell cytosol, thereby enhancing production of the encoded immunogen compared to non-amplifying RNAs (reviewed in [
110,
116]). Due to the robust production of encoded immunogen, saRNAs exhibit the same level of immunogenicity as non-amplifying RNA at a lower dose [
110,
116]. Most saRNAs are developed from positive-sense, single-stranded alphaviruses, such as Venezuelan equine encephalitis virus, Sindbis virus and Semliki forest virus. A simple saRNA, encoding RNA-dependent RNA polymerase and an HIV immunogen, for example, can be delivered to cells through approaches detailed in
Section 3.2. Alternatively, saRNAs can be delivered as viral replicon particles (VRPs). To generate VRPs, the genetic information for alphavirus structural proteins is replaced with HIV antigen sequences. Supplying the recombinant alphavirus structural proteins in-trans to cell culture results in the packaging of VRPs. Without the genetic information for the structural proteins, VRPs are not capable of generating new infectious viral particles [
116]. Several HIV vaccine candidates employing saRNA have been developed and are in testing () [
117,
118] and are discussed in more detail in
Section 3.2.
1.3. Nucleoside Modification of mRNA
An ongoing discussion for mRNA vaccination centers on the use of nucleoside modification to minimize recognition of vaccine mRNAs by innate immune sensors [
9]. RNA binds to and activates toll-like receptor (TLR) 3, TLR7 and TLR8 on DCs, leading to DC maturation and secretion of anti-viral cytokines, especially type I interferons (IFNs) [
119]. Other intracellular virus detection systems, such as the retinoic acid-inducible gene I (RIG-I) and the RIG-I-like receptor (RLR) protein family, RNA-dependent protein kinase (PKR) and the 2-5A system, recognize foreign cytoplasmic RNA and also trigger type I IFN responses, repress RNA translation and promote RNA degradation [
120,
121]. On the one hand, mRNA-induced anti-viral responses may serve to bridge the innate and adaptive immune responses, thus providing a ”self-adjuvating” effect for mRNA vaccines [
122,
123]. Conversely, anti-viral IFN responses have been implicated as inhibitors of vaccine-induced antibody and T cell responses [
124]. We suspect that the timing and/or magnitude of mRNA-induced innate immune responses has important effects on vaccine-induced antibody and T cell responses and is worth further investigation.
For mRNA vaccines in which excessive innate immune activation is undesired, several chemical approaches have been developed to reduce anti-viral responses to vaccine mRNAs. mRNA with modified nucleosides, such as pseudouridine or methylated nucleosides, exhibits much less activation of TLR3, TLR7 and TLR8 than unmodified mRNA [
119]. As a bonus, mRNA containing pseudouridine exhibits enhanced translation compared to unmodified mRNA due to less activation of RNase L and translation suppressor downstream of the 2-5A pathway [
121,
125,
126].
The safety and translational efficiency of vaccine mRNAs can be further optimized by high performance liquid chromatography (HPLC) purification to eliminate double-stranded RNAs, which activate TLR3 [
127,
128]. Strikingly, HPLC-purified mRNA containing pseudouridine induced even lower levels of pro-inflammatory cytokine secretion compared to unpurified, pseudouridine-modified mRNA in DCs, while the translation level was 10- to 1000-fold higher than unpurified mRNA [
128].
2. Progress in mRNA Delivery Strategies for HIV Vaccines
The lack of efficient delivery technologies for mRNA molecules was a major bottleneck to the development of mRNA-based vaccines. The ideal delivery method will protect mRNA from degradation and facilitate cellular entry with minimal toxicity [
8]. mRNA delivery methods that have been tested for HIV vaccine are detailed below and summarized in .
2.1. Electroporation
Electroporation was among the first techniques developed for the delivery of mRNA vaccines. Cu et al. used mice to show that in situ electroporation delivery of saRNA encoding HIV gp140 Env induced higher Env-specific IgG titers and higher frequencies of Env-specific CD4
+ and CD8
+ T cells than naked saRNA [
107]. While the direct application of electroporation to patients is not foreseeable, this technique can be exploited to deliver mRNAs into patient-derived DCs for re-infusion (detailed in
Section 3.2.6).
2.2. Cationic Micelles
Cationic micelles comprising a polyethylenimine-stearic acid copolymer (PSA) were first developed for peptide and protein delivery but were later tested for the delivery of a HIV mRNA vaccine [
129,
130]. PSA molecules self-assemble into nanoparticles in the aqueous phase, with a hydrophobic core formed by stearic acid and a cationic hydrophilic outer layer formed by polyethylenimine, which enables condensation of anionic mRNA molecules. Cationic micelles efficiently deliver mRNAs to DCs with minimal cell death when used at the proper PSA/mRNA ratio and induce DC maturation ex vivo [
130].
To test the efficacy of cationic micelles for HIV mRNA vaccination, mice were immunized with PSA cationic micelles containing non-amplifying mRNA encoding HIV Gag (PSA/mGag) [
130]. PSA/mGag immunization induced higher Gag-specific IgG titers than that elicited by naked Gag mRNA and generated Gag-specific IL-4-secreting CD4
+ T cells and IFN-γ-secreting CD8
+ T cells.
Cationic micelles have also been tested as a means to deliver mRNAs to mucosal sites, an important consideration for HIV vaccination. Cationic micelles consisting of cyclodextrin-polyethylenimine were employed to deliver HIV gp120 mRNA as an intranasal vaccine to mice [
131]. This cationic micelle formulation was able to penetrate epithelial barriers and gp120-specific antibody and T cell responses were evident after two immunizations. Importantly, gp120-specific IgA was detected in both nasal and vaginal washes, indicative of a systemic antibody response that protects distal mucosal sites, an important consideration for sexually transmitted pathogens like HIV.
2.3. Cationic Nanoemulsion
Cationic nanoemulsion (CNE) was developed as a delivery system for saRNA vaccines based on the oil-in-water adjuvant MF59 [
132]. CNE is prepared by mixing Tween 80 with the surfactant Span 85, the cationic lipid DOTAP and squalene. Squalene forms the core of the particle, surfactant stabilizes the emulsion and cationic lipid enables mRNA binding.
In vitro studies demonstrated that CNE protects mRNA from RNase degradation [
132]. saRNA encoding HIV gp140 Env trimer delivered using CNE was compared to three other vaccine platforms in rabbits: saRNA delivery using viral replicon particle (VRP), gp140 Env trimer protein adjuvanted with MF59 and CNE-formulated plasmid DNA [
132]. After two immunizations, a low dose of CNE-formulated saRNA elicited higher Env-specific serum IgG titers than those induced by VRP-delivered RNA or MF59-adjuvanted Env protein. Additionally, all animals immunized with CNE-formulated saRNA exhibited higher neutralization titers against tier 1 (easy to neutralize) pseudoviruses.
In rhesus macaques, the CNE-delivered gp140 saRNA vaccine elicited antibodies that neutralize autologous viruses to a greater degree than VRP-based vaccine but less than immunization with gp140 protein adjuvanted with MF59 [
117]. Interestingly, macaques primed with the CNE-based vaccine then boosted with the gp140 protein in MF59 mounted equivalent antibody responses to macaques primed and boosted with gp140/MF59. Moreover, the Env-specific T cell response to the CNE-containing vaccine was characterized by IFNγ, IL-2 and IL-4 production, whereas VRP and protein subunit vaccines only induced IL-4.
2.4. Poly (lactic acid) Nanoparticle with Cell-Penetrating Peptides
Poly (lactic acid) (PLA) polymer is a biodegradable and biocompatible material that has been extensively studied and approved by the US Food and Drug Administration [
133,
134]. PLA nanoparticles (PLA-NPs) are able to encapsulate a wide variety of proteins and are efficiently taken up by DCs, thus providing a potential system to deliver mRNAs. However, both PLA-NPs and mRNA molecules are hydrophobic and negatively charged, making it impossible for PLA-NPs to encapsulate mRNAs in the core or carry mRNAs directly on the surface. Thus, PLA-NP with cell penetrating peptides (CPPs) have been developed and tested, where cationic CPPs are used as an intermediate to carry mRNAs on PLA-NPs. In in vitro studies, nanocomplexes of PLA-NP/CPP and HIV Gag mRNA were taken up by monocyte-derived DCs which, in turn, expressed Gag protein, underwent DC maturation and secreted pro-inflammatory cytokines. DC maturation was associated with mRNA-dependent TLR3 and RIG-I signaling [
134]. This delivery mechanism is novel and, to our knowledge, in vivo studies using this platform for HIV mRNA vaccination have not been published.
2.5. Cationic Lipid Nanoparticle
Cationic lipid nanoparticles (LNP) consist of a lipid bilayer and typically one or more of the following components: polyethylene glycol (PEG), cholesterol and phospholipid [
8]. They were first developed to deliver pharmaceutical small interfering RNAs (siRNAs) and entered human clinical trials [
135]. LNP exhibit persistent in vivo activity, require lower and less frequent doses compared to other non-viral delivery methods [
136]. Excitingly, the safety and immunogenicity of the mRNA-LNP platform in people has been demonstrated by the two SARS-CoV-2 vaccines, mRNA-1273 and BNT162b2 [
114,
115,
137].
Nucleoside-modified, HPLC-purified mRNAs encoding HIV antigens encapsulated in LNP (mRNA-LNP) have been studied in small animal models and NHPs. In mice, an mRNA-LNP vaccine encoding HIV Env induced robust CD8
+ and CD4
+ T cell responses [
8]. Moreover, in rabbits, vaccination with mRNA-LNP encoding Env gp120 generated anti-gp120 IgG titers six weeks after a single immunization and were further boosted by subsequent immunization [
138]. Virus neutralization activity against tier 1 virus and antibody-dependent cellular cytotoxicity (ADCC) were detected in all rabbits after two immunizations. Similar immune responses were observed in rhesus macaques. Strikingly, 50% of the rhesus macaques also generated autologous tier 2 (difficult-to-neutralize) virus neutralization in the serum.
As discussed in
Section 2, the induction of HIV bnAbs requires robust and persistent germinal centers which facilitate the maturation of bnAb precursors to full-fledged bnAbs. The ability of mRNA-LNP vaccines to induce such robust humoral response to HIV Env is related to its ability to activate TFH cell response. For example, vaccination of rhesus macaques with mRNA-LNP encoding Env induced robust Env-specific TFH cells, whereas there was little evidence of TFH activity following immunization with a protein Env adjuvanted with the TLR3 agonist poly-ICLC [
139]. Thus, the mRNA-LNP platform appears to elicit potent humoral and cellular immune responses to HIV Env.
The mRNA-LNP platform has also been used to deliver bnAbs for passive immunization for HIV [
140]. Humanized mice were injected with mRNA-LNP encoding the heavy and light chains of a CD4 binding site bnAb or with the protein antibody. Interestingly, plasma bnAb levels were higher in mRNA-LNP-injected mice than in the mice receiving a large bolus of antibody as protein, indicating that mRNA-LNP offers a strategy to maintain high concentrations of bnAb in vivo. Most importantly, humanized mice administered bnAb-encoding mRNA-LNP were protected from challenge with HIV.
The molecular mechanisms behind the remarkable immunogenicity of mRNA-LNP vaccines are still being untangled but one mystery is the apparent lack of adjuvant. To our knowledge, vaccine studies with mRNA-LNP have all utilized nucleoside-modified, HPLC purified mRNAs, which lack the “self-adjuvanting” properties of unmodified mRNAs and lipid nanoparticles that do not contain any obvious immunostimulants. A clue was provided by Pardi et al., who injected mice with a mixture of influenza HA protein with mRNA-LNP encoding luciferase and observed a robust anti-HA antibody response, suggesting that the LNPs have inherent adjuvant properties [
139].
2.6. Ex Vivo Loading of Dendritic Cell
So far, we have described mRNA delivery methods in which vaccine recipients are injected with mRNAs (usually in some delivery system), with the hope that some mRNA-encoded immunogens are picked up–or even better, expressed–by DCs. An alternative approach to HIV vaccination involves the removal of monocytes from an individual patient, maturing the monocytes into DCs ex vivo, loading the DCs with mRNA encoding HIV immunogens and then re-infusing the immunogen-loaded DCs back into the patient. This strategy is certainly more labor-intensive and expensive than direct injection of mRNAs but it ensures that DCs are presenting immunogen-derived peptides to activate cognate CD4
+ and CD8
+ T cells. Although DCs are able to endocytose naked mRNAs [
8], other methods, such as electroporation, are typically used to transfect mRNA more efficiently into DCs. The ability of mRNA-transfected DCs to induce potent antigen-specific CD4
+ and CD8
+ T cell responses was demonstrated as early as 2000 with mRNA encoding HIV Gag [
141].
To date, most efforts surrounding ex vivo DC loading and HIV vaccination have focused on their potential as a therapeutic vaccine for HIV-infected individuals. Transfection of mRNA encoding HIV genes into DCs confers the ability to activate CD4
+ and CD8
+ T cells ex vivo [
141,
142,
143] and multiple clinical trials have been conducted to evaluate ex vivo loaded DCs for their ability to control HIV [
144,
145,
146,
147,
148]. In most cases, antigen-specific T cell responses were observed, at least transiently but there was little evidence that any of the therapeutic vaccines controlled viremia. While optimistic about the results, researchers in this field continue to optimize the mRNA-DC vaccine strategy to prolong immune responses.
One compelling mRNA vaccine candidate exploits the capacity of mRNA to encode immunogens as well as immunomodulatory factors. “TriMix” comprises mRNAs encoding three DC activating proteins, CD40L, CD70 and a constitutively-active form of TLR4, which is then mixed with mRNAs encoding conserved HIV peptide sequences from Gag, Pol, Vif and Nef [HIVACAT T-cell immunogen (HTI)]. A murine form of the TriMix-HTI mRNA vaccine induced systemic antigen-specific CTLs after direct injection into the lymph nodes of mice and the human version of the mRNA vaccine induced monocyte-derived DCs to mature and induce T cell proliferation and cytokines ex vivo [
149]. TriMix-HTI was evaluated as a therapeutic vaccine in ART-treated HIV infected individuals in a Phase IIa clinical trial [
150]. Patients were vaccinated with three doses of naked HTI-TriMix, TriMix mRNAs or placebo followed by ART interruption. The vaccine candidate was proved safe and well-tolerated in humans. However, no efficacy in controlling viral rebound was observed. Unfortunately, an extra start codon was mistakenly included in the DNA plasmid template for the mRNA encoding HIV-specific peptides, which may have affected the expression of the immunogen peptides.
Although ex vivo loading of DCs was the first to be evaluated in human clinical trials for HIV mRNA vaccine, this method involves working with autologous DCs, which requires much expertise in cell therapeutics and is laborious and expensive [
122]. The research focus has mostly switched to other chemical, synthetic delivery vehicles.
This entry is adapted from the peer-reviewed paper 10.3390/vaccines9020134