Many authors suggested that IgA Vasculitis (IgAV) and IgA Nephropathy (IgAN) would be two clinical manifestations of the same disease; in particular, that IgAV would be the systemic form of the IgAN. A limited number of studies have included sufficient children or adults with IgAN or IgAV (with or without nephropathy) and followed long enough to conclude on differences or similarities in terms of clinical, biological or histological presentation, physiopathology, genetics or prognosis. All therapeutic trials available on IgAN excluded patients with vasculitis. IgAV and IgAN could represent different extremities of a continuous spectrum of the same disease. Due to skin rash, patients with IgAV are diagnosed precociously. Conversely, because of the absence of any clinical signs, a renal biopsy is practiced for patients with an IgAN to confirm nephropathy at any time of the evolution of the disease, which could explain the frequent chronic lesions at diagnosis.
- IgA Vasculitis
- IgA Nephropathy
- adults
- children
- presentation
- physiopathology
- genetics
- prognosis
- treatment
1. Epidemiology
Apart from the obvious differences in extra-renal clinical presentation, the main difference between IgAV and IgAN is epidemiological (Table 1). The overall precise prevalence of IgAN or IgAV, in children or adults, is not known, but varies considerably around the world [1].
Table 1. Differences and similarities between IgAN and IgAV.
| IgA Nephropathy | IgA Vasculitis | |
|---|---|---|
| Age at onset | 30 to 39 years | 1 to 19 years and 60 to 69 years |
| Clinical presentation | Only renal | Extra-renal symptoms (skin, gastro-intestinal, joint, neurologic, pulmonary, urologic) ± renal involvement |
| Renal biopsy | Mesangial IgA1, IgG, IgM, C3 and fibrin on immunofluorescence Mesangial hyper-cellularity with increased mesangial matrix, endo-capillary hyper-cellularity, segmental glomerular sclerosis, cellular crescents on light microscopy |
|
| Outcome | More severe in adults | |
| Physiopathology | Multi-hit model involving IgA1 | |
In children, IgAV incidence is about 3 to 26.7/100,000 children per year [2][3][4]. The risk of progression to end-stage renal disease requiring dialysis in children varies from 2.5% to 25%, but it is on average around 8% [5][6]. In Europe, 3% of children on dialysis are due to IgAV. It is much rarer in adults, where its incidence is around 1.4 to 5.1/100,000 [7][8][9][10]. The child/adult ratio would therefore vary from 150 to 205. In adults, the risk of developing chronic renal failure is frequent, from 8 to 68%, on average around 18% within 15 years [11][12].
In children, IgAN incidence varies from 0.03/100,000/year in Venezuela to 9.9/100,000/year in Japan and in adults around 2.5/100,000/year [13][14]. The reported incidence rates are likely to underestimate true rates of IgAN, as this disease can exist sub-clinically and depends on country policy for microscopic hematuria detection in the population and/or renal biopsy indications. The risk of progression to end-stage renal disease (ESRD) is reported from 4% after 4.6 years in Europe [15] to 11% after 15 years of follow-up in Japan [16] and 14% in China [17]. The risk of progression to ESRD requiring dialysis in adults varies from 30% to 40% within 10 to 25 years [18]. In adults, IgAN is the cause of ESRD of 3.6% of newly dialyzed patients [19].
Whatever the studies, there is definitely an age difference between patients with IgAN and those with IgAV. This is particularly demonstrated by this Japanese study [20]: Among 18,967 patients with biopsy-proven disease between July 2007 and December 2012, the authors selected 513 patients with IgAV and 5679 with IgAN from the J-RBR (Japan Renal Biopsy registry) registry. They highlight a bimodal distribution for IgAV with two peaks between 1 to 19 years and 60 to 69 years, whereas for IgAN, the peak is rather between 30 to 39 years.
2. Clinical Presentation and Outcome
Macroscopic hematuria is the most frequent IgAN initial presentation in children, followed by the fortuitous finding of microscopic hematuria accompanied or not by proteinuria. In adults, the diagnosis is often made at the stage of chronic kidney disease (CKD), as if diagnosis in this belated context were made at an advanced stage after missing a pauci-symptomatic form that previously occurred in childhood. By contrast, the diagnosis of IgAV is, in most cases, made much earlier, revealed by the presence of extra-renal signs.
Indeed, by definition, the IgAV is characterized by the combination of cutaneous (palpable purpura), gastrointestinal (colicky pain, bloody stools) and articular (arthralgia) involvement. More rarely, we can observe a neurological, pulmonary or urological involvement [21]. The long-term prognosis of the disease depends on the presence of renal impairment and its evolution. From a histological point of view, it is not possible to distinguish a glomerulonephritis as part of an IgAV from an IgAN. Renal biopsy shows, in the two cases: on immunofluorescence, predominant IgA1 deposits in the mesangium of all glomeruli (Figure 1), with glomerular deposits of IgG, IgM, C3 and fibrin in variable proportions; on light microscopy, mesangial hypercellularity with increased mesangial matrix, endo-capillary hypercellularity, segmental glomerular sclerosis, cellular crescents and tubular atrophy and interstitial fibrosis.
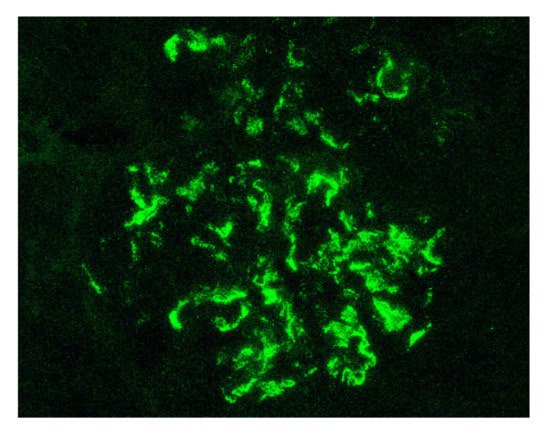
Figure 1. Mesangial and capillary wall IgA deposits (immunofluorescence staining for IgA, original magnification ×400).
IgAV is most common in childhood, but may occur at any age. Macroscopic hematuria is very uncommon after age 40. The importance of asymptomatic urine abnormality as the presentation of IgAN will depend on attitudes to routine urine testing and renal biopsy. It is unclear whether patients referring late, with chronic renal impairment, have a disease distinct from those referring earlier, with macroscopic hematuria (Figure 2: Data from patients presentation in Leicester, UK, 1980–1995 [22]).
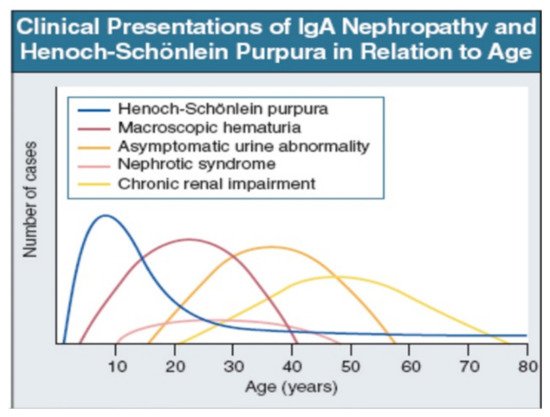
Figure 2. IgAN and IgAV clinical presentation over time from childhood to older age [22]. Reprinted with permission from ref. [22]. Copyright 2000 Rights and Permissions of Elsevier.
Renal involvement occurs in IgAV with a prevalence ranging from 20 to 54% in children and 45 to 85% in adults. Patients with nephritis have hematuria ± proteinuria of varying flow rate. They seem to have a higher frequency of nephritis and rapidly progressive nephritic syndrome at onset, notably in adults, but this is not the case in all published cohorts, as some of them, as we will develop furthermore, even describe the opposite.
Whether or not disease severity at diagnosis and prognosis differs between IgAN and IgAV remains controversial.
With regard to the clinical presentation and prognosis, the results of the few studies comparing the two diseases most frequently show a less favorable evolution for IgAN. Significant differences in the conclusions of the studies from one country to another suggest genetic susceptibility.
A Chinese study [23] compared 31 children with IgA Nephropathy to 120 children with IgA vasculitis and nephropathy, 32 of whom had a renal biopsy. In this pediatric cohort, patients with IgAN were significantly older. Histologically, the kidneys of patients with IgAN were more sclerotic (35.5% versus 3.1%) while there were more endothelial proliferation in those with IgAV (65.6% versus 29%). After 34 months of follow-up, the prognosis seemed better in patients with IgAV, since 72.5% of children from the IgAV group were in complete remission compared with only 19.4% in the IgAN group, in which patients had significantly persistent hematuria and proteinuria.
In another Spanish study [24], 142 patients with IgAV were compared to 61 patients with IgAN, of all ages. Those with IgAV were also younger (mean age at onset 30.6 years (2.9 to 82.7 years), than those with IgAN (37.1 years (14.7 to 78.5 years)). Again, the renal presentation was less severe: a lower rate of renal insufficiency (25% in IgAV vs. 63.4% in IgAN) and nephrotic syndrome (12.5% vs. 43.7%) were reported, while the prognosis, after a median follow-up of 130 months, was better: dialysis (2.9% in IgAV vs. 43.5% in IgAN), renal transplant (0% vs. 36%) and residual chronic renal insufficiency (4.9% vs. 63.8%)
In a Korean study [25], 92 adult patients with kidney biopsy-proven IgAV were compared to 1011 adult patients with kidney biopsy-proven IgAN, 89 and 178, respectively, from each group were matched using a propensity score in order to compare long term renal outcome. Once again patients with nephropathy in the context of IgAV were younger (33.2 ± 15.9 years vs. 37.7 ± 13 years), and less frequently developed renal insufficiency (7.6% vs. 14.4%) and interstitial fibrotic lesions on biopsy: however, these differences disappeared once the propensity score was applied to the subgroup. In addition, while the renal prognosis seemed better in the IgAV group (ESRD 14.1% vs. 22%; remission 28.3% vs. 13.8%), this difference disappeared (ESRD 14.6 vs. 13.5%; remission 28.1% vs. 27.5%) in the cohort of patients matched for clinical, histological and treatment factors. In my opinion, this study is essential, and it is the one that more clearly sheds light on the matter. It substantiates that the worse prognosis ascribed, from the other above-mentioned studies, to IgA Nephropathy, is probably pointed out only because we selected the most severe patients, namely, those who had not recovered spontaneously.
A Chinese pediatric study [26] compared the clinical-histological presentation of 41 patients with IgAN and 137 with IgAV. Patients with IgAV had higher levels of blood white cell, hemoglobin and platelet, and lower levels of hematuria, blood nitrogen and C4, compared to IgAN cases, but without any clinical and histological renal significant difference. The authors did not perform any prospective follow-up.
Another Japanese study [20] compared 513 patients with IgAV and 5679 with IgAN, of all ages, from a renal national biopsy registry. As previously mentioned, the age at diagnosis considerably differed: IgAV peaked at 1 to 19 years and 60 to 69 years, whereas IgAN peaked at 30 to 39 years. In contrast to previous studies, it appears that, regardless of age, patients with IgAV had a more severe clinical presentation with more frequent rapidly progressive renal insufficiency (4.5% vs. 1.4%) and a nephrotic syndrome (10.5% vs. 3%), and biopsies showed more inflammatory lesions including more endocapillary proliferation (6.4% vs. 0.9%) and extra-capillary (6.6% vs. 0.8%) glomerulonephritis. In contrast, patients with IgAN, had significantly more chronic nephritic syndrome (88.5% vs. 61.6%). Unfortunately, this cross-sectional study had no reference to the renal outcomes for any of the patients.
A further Japanese study [27] analyzed cross-sectionally 24 IgAV adult patients with nephritis or 56 adult patients with IgAN, all of whom underwent renal biopsy. The clinical characteristics did not differ between groups. Duration from onset was significantly longer for IgAN (47.6 months vs. 8.8 months). More patients with IgAV received steroid therapy, whereas more patients with IgAN received renin-angiotensin-aldosterone system (RAAS) blockers. Compared to IgAN, the mean rate of global sclerosis or crescent formation were significantly lower in patients with IgAV. Using Oxford classification, IgAV patients had more endothelial injury and IgAN worse mesangial proliferation, crescent formation, and tubulointerstitial injury.
A French retrospective study [28] showed that the risk score of end-stage renal disease or death, including hypertension, proteinuria more than 1 gramme per day and severe pathological lesions (local classification score ≥ 8), validated in a cohort of 1064 patients with an IgAN, was also valid when it was applied to a subgroup of 74 patients with IgAV from this same cohort, after 8.2 years of follow-up.
More recently, the National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) funded the CureGN study to establish a primary glomerular disease consortium with a focus on IgAN and IgAV. Using this longitudinal observational cohort of adults and children with biopsy-proven primary glomerular disease, from all over the world, this first report described the baseline clinical characteristics of the two nosological entities [29]. The next step will be to better predict the long-term follow-up renal outcome, find prognostic biomarkers and identify patients most appropriate for specific therapies. A total of 667 patients were enrolled, including 506 (75.9%) with IgAN and 161 (24.1%) with IgAV, 285 (42.7%) were children. At the moment of biopsy, patients with IgAV were younger, more frequently white, and had a higher estimated glomerular filtration rate and lower serum albumin than those with IgAN. Adults and children with IgAV were similar in terms of proteinuria, hematuria or serum albumin. Patients with IgAV were more likely to be treated with immunosuppressive therapy if compared to those with IgAN, but less likely to receive standard supportive care with RAAS inhibition. No data is available to date about renal histology.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10112310
References
- O’Shaughnessy, M.M.; Hogan, S.L.; Thompson, B.D.; Coppo, R.; Fogo, A.B.; Jennette, J.C. Glomerular Disease Frequencies by Race, Sex and Region: Results from the International Kidney Biopsy Survey. Nephrol. Dial. Transplantat. 2018, 33, 661–669.
- Gardner-Medwin, J.M.; Dolezalova, P.; Cummins, C.; Southwood, T.R. Incidence of Henoch-Schonlein Purpura, Kawasaki Disease, and Rare Vasculitides in Children of Different Ethnic Origins. Lancet 2002, 360, 1197–1202.
- Piram, M.; Maldini, C.; Biscardi, S.; De Suremain, N.; Orzechowski, C.; Georget, E.; Regnard, D.; Koné-Paut, I.; Mahr, A. Incidence of IgA vasculitis in children estimated by four-source capture–recapture analysis: A population-based study. Rheumatology 2017, 56, 1358–1366.
- Mossberg, M.; Segelmark, M.; Kahn, R.; Englund, M.; Mohammad, A. Epidemiology of Primary Systemic Vasculitis in Children: A Population-Based Study from Southern Sweden. Scand. J. Rheumatol. 2018, 47, 295–302.
- Yang, Y.H.; Hung, C.F.; Hsu, C.R.; Wang, L.C.; Chuang, Y.H.; Lin, Y.T.; Chiang, B.L. A Nationwide Survey on Epidemiological Characteristics of Childhood Henoch-Schonlein Purpura in Taiwan. Rheumatology 2005, 44, 618–622.
- Okubo, Y.; Nochioka, K.; Sakakibara, H.; Hataya, H.; Terakawa, T.; Testa, M.; Sundel, R.P. Nationwide epidemiological survey of childhood IgA vasculitis associated hospitalization in the USA. Clin. Rheumatol. 2016, 35, 2749–2756.
- Watts, R.A.; Lane, S.; Scott, D.G. What is known about the epidemiology of the vasculitides? Best Pract. Res. Clin. Rheumatol. 2005, 19, 191–207.
- Penny, K.; Fleming, M.; Kazmierczak, D.; Thomas, A. An epidemiological study of henoch-schonlein purpura. Paediatr. Nurs. 2010, 22, 30–35.
- Romero-Gómez, C.; Aguilar-García, J.A.; García-de-Lucas, M.D.; Cotos-Canca, R.; Olalla-Sierra, J.; García-Alegría, J.J.; Hernández-Rodríguez, J. Epidemiological study of primary systemic vasculitides among adults in southern spain and review of the main epidemiological studies. Clin. Exp. Rheumatol. 2015, 33, S-11–S-18.
- Hočevar, A.; Rotar, Z.; Ostrovršnik, J.; Jurčić, V.; Vizjak, A.; Dolenc Voljč, M.; Lindič, J.; Tomšič, M. Incidence of IgA Vasculitis in the Adult Slovenian Population. Br. J. Dermatol. 2014, 171, 524–527.
- Pillebout, E.; Thervet, E.; Hill, G.; Alberti, C.; Vanhille, P.; Nochy, D. Henoch-schonlein purpura in adults: Outcome and prognostic factors. J. Am. Soc. Nephrol. 2002, 13, 1271–1278.
- Huang, X.; Wu, X.; Le, W.; Hao, Y.; Wu, J.; Zeng, C.; Liu, Z.; Tang, Z. Renal prognosis and related risk factors for henoch-schönlein purpura nephritis: A Chinese adult patient cohort. Sci. Rep. 2018, 8, 5585.
- McGrogan, A.; Franssen, C.F.M.; de Vries, C.S. The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol. Dial. Transplantat. 2011, 26, 414–430.
- Shibano, T.; Takagi, N.; Maekawa, K.; Mae, H.; Hattori, M.; Takeshima, Y.; Tanizawa, T. Epidemiological Survey and Clinical Investigation of Pediatric IgA Nephropathy. Clin. Exp. Nephrol. 2016, 20, 111–117.
- Coppo, R. Pediatric IgA Nephropathy in Europe. Kidney Dis. 2019, 5, 182–188.
- Yoshikawa, N.; Tanaka, R.; Iijima, K. Pathophysiology and Treatment of IgA Nephropathy in Children. Pediatr. Nephrol. 2001, 16, 446–457.
- Wu, H.; Fang, X.; Xia, Z.; Gao, C.; Peng, Y.; Li, X.; Zhang, P.; Kuang, Q.; Wang, R.; Wang, M. Long-term renal survival and undetected risk factors of IgA nephropathy in Chinese Children—a Retrospective 1243 cases analysis from single centre experience. J. Nephrol. 2020, 33, 1263–1273.
- Magistroni, R.; D’Agati, V.; Appel, G.; Kiryluk, K. New developments in the genetics, pathogenesis, and therapy of IgA Nephropathy. Kidney Int. 2015, 88, 974–989.
- Lassalle, M.; Couchoud, C.; Prada-Bordenave, E.; Jacquelinet, C. Éditorial. Néphrol. Thér. 2013, 9, S1.
- Komatsu, H.; Fujimoto, S.; Yoshikawa, N.; Kitamura, H.; Sugiyama, H.; Yokoyama, H. Clinical manifestations of henoch-schonlein purpura nephritis and iga nephropathy: Comparative analysis of data from the Japan Renal Biopsy Registry (J-RBR). Clin. Exp. Nephrol. 2016, 20, 552–560.
- Ozen, S.; Pistorio, A.; Iusan, S.M.; Bakkaloglu, A.; Herlin, T.; Brik, R.; Buoncompagni, A.; Lazar, C.; Bilge, I.; Uziel, Y.; et al. EULAR/PRINTO/PRES criteria for henoch-schonlein purpura, childhood polyarteritis nodosa, childhood wegener granulomatosis and childhood takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann. Rheum. Dis. 2010, 69, 798–806.
- Comprehensive Clinical Nephrology, 6th ed.; Feehally, J. (Ed.) Elsevier: Edinburgh, UK ; New York, NY, USA, 2019; ISBN 978-0-323-47909-7.
- Zhou, J.; Huang, A.; Liu, T.; Kuang, Y. A clinico-pathological study comparing Henoch-Schonlein purpura nephritis with IgA nephropathy in children. Zhonghua Er Ke Za Zhi 2003, 41, 808–812.
- Calvo-Rio, V.; Loricera, J.; Martin, L.; Ortiz-Sanjuan, F.; Alvarez, L.; Gonzalez-Vela, M.C.; Gonzalez-Lamuno, D.; Mata, C.; Gortazar, P.; Rueda-Gotor, J.; et al. Henoch-schonlein purpura nephritis and IgA nephropathy: A comparative clinical study. Clin. Exp. Rheumatol. 2013, 31, S45–S51.
- Oh, H.J.; Ahn, S.V.; Yoo, D.E.; Kim, S.J.; Shin, D.H.; Lee, M.J.; Kim, H.R.; Park, J.T.; Yoo, T.H.; Kang, S.W.; et al. Clinical outcomes, when matched at presentation, do not vary between adult-onset henoch-schonlein purpura nephritis and IgA nephropathy. Kidney Int. 2012, 82, 1304–1312.
- Mao, S.; Xuan, X.; Sha, Y.; Zhao, S.; Zhu, C.; Zhang, A.; Huang, S. Clinico-pathological association of henoch-schoenlein purpura nephritis and IgA nephropathy in children. Int. J. Clin. Exp. Pathol. 2015, 8, 2334–2342.
- Sugiyama, M.; Wada, Y.; Kanazawa, N.; Tachibana, S.; Suzuki, T.; Matsumoto, K.; Iyoda, M.; Honda, H.; Shibata, T. A cross-sectional analysis of clinicopathologic similarities and differences between henoch-schönlein purpura nephritis and IgA nephropathy. PLoS ONE 2020, 15, e0232194.
- Mohey, H.; Laurent, B.; Mariat, C.; Berthoux, F. Validation of the absolute renal risk of dialysis/death in adults with IgA nephropathy secondary to henoch-schönlein purpura: A monocentric cohort study. BMC Nephrol. 2013, 14, 169.
- Selewski, D.T.; Ambruzs, J.M.; Appel, G.B.; Bomback, A.S.; Matar, R.B.; Cai, Y.; Cattran, D.C.; Chishti, A.S.; D’Agati, V.D.; D’Alessandri-Silva, C.J.; et al. Clinical characteristics and treatment patterns of children and adults with IgA nephropathy or IgA vasculitis: Findings from the cureGN study. Kidney Int. Rep. 2018, 3, 1373–1384.