With advancing aging, a decline in physical abilities occurs, leading to reduced mobility and loss of independence. Although many factors contribute to the physio-pathological effects of aging, an important event seems to be related to the compromised integrity of the neuromuscular system, which connects the brain and skeletal muscles via motoneurons and the neuromuscular junctions (NMJs). NMJs undergo severe functional, morphological, and molecular alterations during aging and ultimately degenerate. The effect of this decline is an inexorable decrease in skeletal muscle mass and strength, a condition generally known as sarcopenia. Moreover, several studies have highlighted how the age-related alteration of reactive oxygen species (ROS) homeostasis can contribute to changes in the neuromuscular junction morphology and stability, leading to the reduction in fiber number and innervation. Increasing evidence supports the involvement of epigenetic modifications in age-dependent alterations of the NMJ. In particular, DNA methylation, histone modifications, and miRNA-dependent gene expression represent the major epigenetic mechanisms that play a crucial role in NMJ remodeling. It is established that environmental and lifestyle factors, such as physical exercise and nutrition that are susceptible to change during aging, can modulate epigenetic phenomena and attenuate the age-related NMJs changes.
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
The decline in physical ability and reduced mobility represent hallmarks of aging. While several events are known to contribute to muscle loss during aging, including chronic low-grade inflammatory status [
1], reduced protein synthesis [
2], mitochondrial dysfunction [
3], and impaired satellite cell function [
4], it is generally accepted that the decline in neural function is one of the principal factors leading to the pathological status known as sarcopenia [
5,
6,
7]. Indeed, the nervous system not only controls muscle contraction and voluntary movements [
8] but also affects myoblast orientation [
9], skeletal muscle fiber specification, and myosin heavy chain (MHC) isoform expression [
10]. Skeletal muscle fibers are classified as fast or slow according to the motor neuron activity, their distinct oxidative capacities, and ATPase activities [
11]. These properties may be altered by a decrease in motor units, denervation of muscle fibers, and changes in NMJs [
12]. Indeed, slow muscle phenotype is promoted by tonic motor neuron activity, while infrequent motor neuron firing results in fast fiber generation [
12]. Moreover, cross reinnervation experiments demonstrated that fast muscles switch into slow ones when re-innervated by a slow nerve, whereas slow muscles switch into fast ones once re-innervated by fast nerve [
13,
14]. It has been reported by Klitgaard et al. that co-expression of different isoforms of MHC in a single muscle fiber increases with age, suggesting a continuous shift in the type of muscle fiber [
15], and it is demonstrated that muscles in elderly individuals contain more slow muscle fibers that reduce contraction ability [
16]. This also supports endplate fragmentation and functional denervation [
17].
The inability of aged motor axons to efficiently re-innervate muscle fibers following bouts of degeneration [
18] contributes to the atrophy of muscle fibers and loss of motor neurons. Motor neuron loss accompanied by muscle fiber loss may result in a consequent age-related increase in intermuscular fat and fibrous tissue [
19,
20], a minor degree of muscle fiber grouping [
21], and a decrease in muscle size [
6,
22].
One possible mechanism of this progressive impairment of the re-innervation process seems related to age-associated degeneration of NMJ. NMJ is a synaptic connection where the peripheral nervous system contacts skeletal muscle fibers, thus controlling essential processes such as breathing and body voluntary movements [
8]. Significant deterioration, including postsynaptic apparatus fragmentation, denervation, multiple innervations, and axon sprouting, has been observed in this critical region with advancing age [
23,
24,
25]. Interestingly, similar defects characterize several severe motor neuron diseases such as amyotrophic lateral sclerosis (ALS), suggesting that strategies aimed to preserve NMJ integrity may be crucial in understanding the critical components involved in aging and ALS [
26,
27].
Although skeletal muscle fibers are characterized by having many nuclei, only those placed in the proximity of the NMJ are important for the transcription of the genes involved in NMJ structure, function, and maintenance [
28,
29,
30]. The correct localization of synaptic nuclei at the NMJ requires the interplay between the cytoskeleton and several nuclear envelope proteins to effectively transduce cytoskeletal forces to the nucleus to regulate signaling pathways, chromatin organization, and gene expression [
31,
32,
33,
34]. It has been demonstrated that lamin A/C, an intermediate filament involved in establishing a continuous physical link between the nuclear lamina and the cytoskeleton [
35,
36,
37,
38], is reduced in aged muscles, and its mutations are implicated in premature aging disorders [
39,
40,
41]. In particular, a recent paper published by Gao et al. demonstrated that muscle-specific lamin A/C mutation results in progressive denervation, AChR cluster fragmentation, and neuromuscular dysfunction [
42]. These results demonstrate that lamin A/C is critical in the maintenance of NMJ and suggest that alterations in the cytoskeleton-nuclear network may contribute to NMJ degeneration in aged muscle.
Increasing evidence supports the involvement of epigenetic modifications in age-dependent morphological and functional alterations of the NMJ [
25,
43,
44]. In particular, DNA methylation, histone modifications, and miRNA-dependent gene expression represent the major epigenetic mechanisms that play a crucial role in NMJ remodeling [
45,
46,
47].
2. Age-Related Changes in NMJ
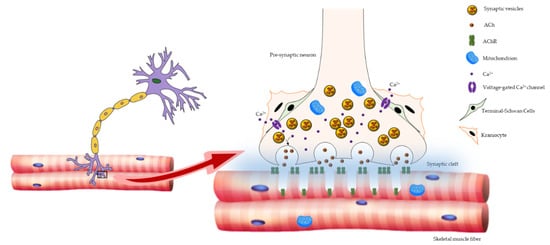
Three principal elements compose NMJ: the presynaptic motor nerve terminals packed with synaptic vesicles containing the neurotransmitter acetylcholine (ACh); the synaptic cleft; and the deeply folded postsynaptic membrane of the muscle fiber, also referred to as the endplate, where acetylcholine receptors (AChRs) are located. When an action potential reaches the pre-synaptic element, voltage-dependent calcium channels open, allowing calcium to trigger the delivery of ACh in the synaptic cleft. Acetylcholine triggers AChR located in the post-synaptic membrane to produce an action potential, which in turn activates voltage-gated dihydropyridine receptors (DHPRs) located in the sarcolemma, and subsequently ryanodine receptors (RyRs) located in the sarcoplasmic reticulum membrane. The nerve terminal is covered by specialized glial cells, called terminal Schwann cells (tSCs), that produce a basal lamina that fuses with that of the muscle fiber at the boundary of the NMJ [
48,
49,
50]. Moreover, fibroblast-like cells, known as kranocytes or perisynaptic fibroblasts, form a loose cover over the NMJ and participate in nerve repair and regeneration, thus representing other crucial players in neuromuscular functionality [
50,
51] ().
Figure 1. Schematic representation of NMJ architecture. NMJ is composed of three major elements: the presynaptic motor nerve terminals packed with synaptic vesicles containing the neurotransmitter ACh; the synaptic cleft; and the deeply folded postsynaptic membrane of the muscle fiber, where AChRs are placed. The NMJ-associated cells, tSCs, and kranocytes are also shown.
During aging, NMJs undergo dramatic morphological, functional, and molecular changes that lead them to ultimately degenerate [
52,
53,
54]. At the level of the presynaptic region, increased axon diameter and larger nerve terminal area have been observed [
23,
55]. However, this effect is not accompanied by increased Ach stores, since it has been demonstrated that as presynaptic branching increases with age, the quantity of available Ach decreases [
56,
57]. At the postsynaptic level, the endplates decrease in size and are severely fragmented, and the number and length of postsynaptic folds are reduced, leading to a functional impairment of NMJ response [
58]. Along with these modifications, a gradual decrease in the number of AChRs per junction has been observed [
59]. Moreover, tSCs migrate away from the motor nerve terminal and protrude branches into the synaptic cleft, contributing to a functional decline of the neuromuscular system in aging [
21,
60].
A key role in the communication between nerve and muscle is played by the agrin-muscle-specific kinase (MuSk)–low-density lipoprotein receptor-related protein 4 (Lrp4) signaling pathway, which ensures the correct transmission of the action potential from the nerve terminal to the endplate, thus facilitating myofibrillar excitation [
61,
62]. It has been demonstrated that the agrin–MuSk–Lrp4 signaling pathway may be dysregulated with advancing aging. Agrin is expressed at NMJs by nerve terminals, muscle fibers, and tSC, where it is necessary for clustering of AchRs in muscle fibers [
63,
64,
65,
66,
67,
68]. In a mouse experimental model, Agrin impaired expression resulted in NMJ fragmentation, similar to what was observed in aged NMJs and premature sarcopenia [
69]. Moreover, it has been demonstrated that the expression levels of NMJ-associated genes MuSk and Lrp4 appear to increase in aged muscle [
70]. Interestingly, these genes are also upregulated in the denervated skeletal muscle of young mice, suggesting that aging may cause denervation of NMJs [
71].
It has also been demonstrated that mitochondria dysfunction and oxidative stress, along with increased levels of intracellular calcium boost, determine the decline in NMJ [
72,
73], together with a reduced number of synaptic vesicles, similarly to what was observed in ALS [
27,
74,
75,
76].
3. ROS and NMJ Degeneration
Reactive oxygen species (ROS) are highly reactive molecules that are generated due to the electron receptivity of O
2. In eukaryotic cells, endogenous production of ROS is associated with mitochondrial oxidative phosphorylation, by which a chain of redox reactions transports protons across the inner mitochondrial membrane, generating potential energy that accomplishes cellular ATP production [
77].
One of the major molecules that play an important role in mitochondrial function is calcium (Ca2+). During muscle contraction, a greater influx of Ca2+ into the mitochondria occurs, which results in the activation of specific enzymes that lead to an increased synthesis of ATP to accomplish the energy demand. Moreover, increased mitochondrial Ca2+ levels have also been observed in several pathological conditions such as skeletal muscle denervation or unloading. The result of this steady-state elevation of mitochondrial Ca2+ level leads to increased ROS generation, induction of programmed cell death, and ultimately muscle atrophy [
78].
In physiological conditions, a small number of electrons, passing through the chain, prematurely and incompletely reduces oxygen, producing the superoxide radicals; these are promptly scavenged by the cellular antioxidant enzymes, such as superoxide dismutase 1 (SOD1), to avoid oxidative damage of cellular macromolecules. Therefore, mitochondrial dysfunction and oxidative phosphorylation impairment determine the increase of ROS production and oxidative damage; these events, in turn, induce muscle cells to respond to the unbalanced redox homeostasis, upregulating the antioxidant enzymes [
79]. Experimental evidence indicates that ROS may also act as chemical messengers, involved in receptor-mediated signaling pathways and transcriptional activation [
80], suggesting that the cellular balance between ROS production and ROS scavenging is crucial to preserve cellular functionality [
81].
Abnormal ROS levels contribute to the etiology of a wide variety of disorders, including aging and neurodegenerative diseases, such as ALS, and most of them are characterized by incoming defects in muscle–nerve communication [
82].
Several animal models have been generated to elucidate the relation between oxidative damage and the alteration of the muscle–nerve endplate; among these are the transgenic mice overexpressing in skeletal muscle tissue the uncoupling protein 1 (UCP1), a mitochondrial protein that uncouples mitochondrial electron transport from ATP synthesis. These mice display deterioration of the NMJ, which correlates with progressive signs of muscle denervation and late-onset motor neuron pathology [
83], demonstrating that a relatively mild mitochondrial dysfunction can recapitulate early signs of ALS disease, and it is sufficient to determine NMJ alteration and motor neuron degeneration.
Several other studies have investigated the relationship between alteration of antioxidant defense and NMJ defects. Data from SOD1 knockout mice (SOD1
−/−) [
84] and mutant SOD1 gain-of-function models (SOD1
G93A mice) [
85,
86] demonstrate the importance of SOD1 enzyme activity and ROS homeostasis in muscle–nerve communication. Indeed, it has been shown that the ubiquitous expression of the mutant human SOD1
G93A gene induces a widespread spinal motor neuron death in mice. Conversely, the genetic ablation of the SOD1 gene (SOD1
−/−) in mouse determines, at advanced ages, a motor neuropathy associated with distal motor axon degeneration, without loss of ventral motor neurons, confirming the crucial role of the SOD1 antioxidant activity in the maintenance of NMJ stability. Interestingly, muscle denervation in SOD1
−/− strongly correlates with loss of mitochondria at the motor nerve terminal during postnatal life. The defect is rescued by the replacement of SOD1 enzyme in the mitochondrial inner membrane [
87], indicating the close relationship between the alteration of mitochondria functionality, induced by oxidative stress, and defects in muscle–nerve communication.
Although it is widely accepted that NMJ alteration during aging or ALS disease is closely related to axon and synapse damage, controversy exists on whether pathological events could begin at the skeletal muscle, potentially influencing the loss of NMJs and the degeneration of motor neurons. Indeed, the temporal analysis of axon and neuromuscular junction degeneration in an ALS mouse model indicated that motor neuron degeneration starts distally at the NMJ area and occurs earlier than clinical symptoms, proceeding towards neuron soma in a retrograde dying back manner [
88].
The transgenic mice MLC/SOD1G93A, overexpressing the SOD1G93A gene under the control of a muscle-specific promoter, provide further evidence to support the dying-back theory and the concept that skeletal muscle is a primary target of the toxic effect of the SOD1 mutant gene. Indeed, the transgenic MLC/SOD1G93A mice exhibit defects of the mitochondrial membrane potential and reduced integrity of the mitochondrial network, which are associated with higher turnover and fragmentation of the NMJ [
75]. This evidence is corroborated by a recent work of Wong and colleagues who demonstrated that skeletal muscle-restricted expression of wild-type or mutant hSOD1 genes causes age-related ALS-like phenotype [
89], that is, motor defects, distal axonopathy, NMJ pathology, and motor neuron loss, according to a non-autonomous mechanism for motor-neuron degeneration [
90].
The important role of the antioxidant enzymes in muscle and nerve communication has been investigated in several studies focused on ROS production during muscle contraction [
91]. Contractile activity in muscles of young rodents leads to the extracellular release of ATP and the activation of an NADPH oxidase (NOX2) complex in the muscle post-synaptic terminal. NOX2 is responsible for superoxide and hydrogen peroxide (H
2O
2) production and regulates, together with ATP, the release of ACh from the motor nerve terminal [
92]. NOX2 activates redox-sensitive transcription factors that in turn induce higher expression of antioxidant enzymes, such as SOD and catalase, to counteract oxidative damage and facilitate exercise-induced muscle tissue remodeling [
93]. It has been reported that exercise, through the activation of mitogen-activated protein kinases (MAPKs) and through nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) signaling pathways, can cause the expression of antioxidant enzymes that play a crucial role in the protection against ROS, as well as adaptation to exercise [
94,
95]. However, the beneficial effects of physical activity are lost with exhaustive endurance and resistance exercise since the increased levels of ROS observed in these conditions overwhelm cellular antioxidant defenses, leading to tissue damage [
96,
97,
98,
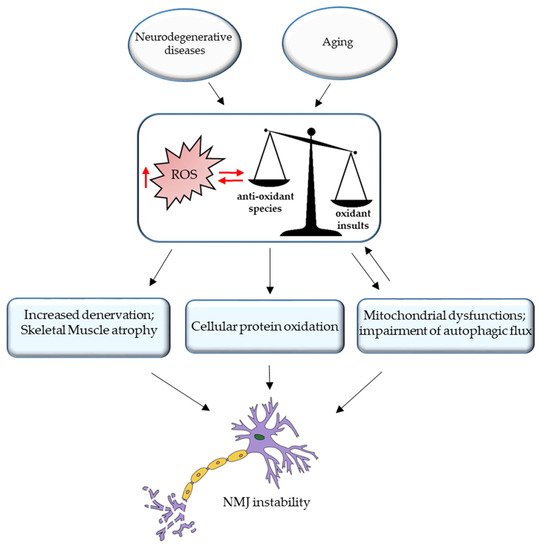
99]. Excessive ROS production during aging or neurodegenerative disorder is responsible not only for mitochondria damage but also for cellular protein oxidation (). Elderly muscles accumulate damaged proteins and organelles, since the activity of the autophagy system, a regulated mechanism for the removal of unnecessary or dysfunctional components, declines during aging. Indeed, in a recent paper [
100], the authors described how the inhibition of the autophagic flux enhances oxidative stress in young muscles and alters muscle force generation, inducing mitochondrial dysfunction and NMJ instability; in this context, autophagy emerges as a required mechanism for mitochondrial quality control and the correct interplay between muscle and nerve [
101].
Figure 2. Oxidative stress and NMJ instability. Excessive ROS production during aging or neurodegenerative disorder is responsible for the increased denervation and skeletal muscle atrophy, cellular protein oxidation, mitochondrial dysfunction, and impairment of autophagic flux, leading to NMJ instability.
In conclusion, this evidence demonstrates that the age-related alteration of ROS homeostasis affects muscle–nerve communication; therefore, the maintenance of redox balance can be considered as a good therapeutic tool to counteract the decline of NMJ functionality of elderly individuals.
This entry is adapted from the peer-reviewed paper 10.3390/cells10061307