Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Nucleoli form around actively transcribed ribosomal RNA (rRNA) genes (rDNA), and the morphology and location of nucleolus-associated genomic domains (NADs) are linked to the RNA Polymerase I (Pol I) transcription status. The number of rDNA repeats (and the proportion of actively transcribed rRNA genes) is variable between cell types, individuals and disease state. Substantial changes in nucleolar morphology and size accompanied by concomitant changes in the Pol I transcription rate have long been documented during normal cell cycle progression, development and malignant transformation.
- nucleolus
- nucleolar associated domain (NAD)
- ribosomal genes
- RNA polymerase I
- transcription
- heterochromatin
- genome architecture
- cell fate
- differentiation
- cancer
1. Nucleoli and the rDNA Genes
Although genetic information is encoded in a linear DNA sequence, the transcription of particular genes (or gene clusters) depends on the surrounding chromatin structure and higher-order chromosomal interactions. Eukaryotic chromatin is tightly packed into the nucleus with a portion compressed into subnuclear domains, one of which is the nucleolus. Nucleoli form around ribosomal RNA (rRNA) genes (rDNA) and dictate the nucleolar morphology and the positioning of nucleolar-associated chromatin domains (NADs) within the nucleus. rRNA genes were first visualized in yeast in the late 1960s by Miller and Beatty using Miller spreads, which provided structural details of actively transcribed rRNA genes, specifically showing a single rDNA repeat transcribed by a multitude of RNA Polymerase I (Pol I) complexes, which they described as the Christmas tree structure [1]. These preparations further revealed that around 100 Pol I molecules simultaneously transcribe one gene at a speed of approximately 95 nucleotides/second [2]. In higher eukaryotes, the presence of histones in the transcribed region is a matter of debate, but it is widely accepted that the transcribed region is deprived of fully assembled nucleosomes [2,3], which are replaced by upstream binding factor (UBTF). The transcribed 47S precursor rRNA (pre-rRNA) is rapidly processed into the mature 28S, 5.8S and 18S rRNA, which assemble together with the 5S rRNA synthesized by RNA Polymerase III and approximately 79 ribosomal proteins translated from mRNAs transcribed by RNA Polymerase II (Pol II) into the 40S and 60S ribosomal subunits. While the process of ribosome biogenesis (RiBi) has long been associated with the nucleolus, more recently other essential non-ribosomal cellular functions have been attributed to this nuclear subdomain. The nucleolus is now recognized as a plurifunctional hub coordinating the nucleolar surveillance pathway in response to cellular stress [4,5,6,7,8], a modulator of genome architecture [9,10,11] and a phase-separated compartment for protein quality control [12].
2. Canonical and Non-Canonical rDNA Repeats
In humans, the rDNA genes are arranged in a head-to-tail orientation forming repeat arrays organized in the nucleolar organizer regions (NOR) at the short arm of the 5 acrocentric chromosomes. The precise organization and exact number of repeats is species, cell type and age dependent [13,14]. Canonical repeats in human cells are 43–45 kb in length and composed of a core and spacer promoter, a transcribed region containing both a 5′ and 3′ external transcribed spacer region (ETS), two internal transcribed spacer regions (ITS) and the 18S, 5.8S and 28S rRNA coding regions, with each individual repeat separated by a non-coding intergenic spacer (IGS; ~30 kb) [15] (Figure 1). In higher eukaryotes, the core promoter dictates transcription of the pre-47S rRNA [16,17], whereas the spacer promoter [18,19,20,21] mediates transcription of non-coding RNAs (see below). Transcription termination factor 1 (TTF-1) binds to the transcription terminator sites downstream of the 28S coding region and blocks Pol I elongation [22,23,24].
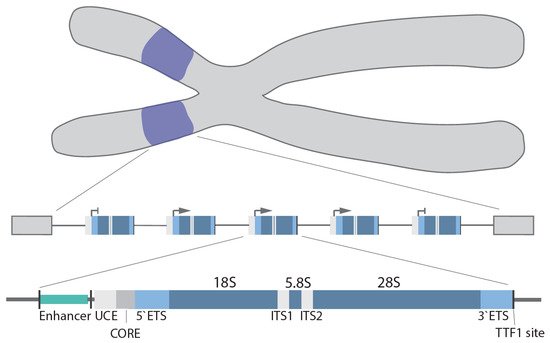
Figure 1. rDNA gene arrays (purple) are located on the short arms of the human acrocentric chromosomes. Organization of a single rDNA gene: enhancer, upstream control element (UCE), core promoter (CORE), 5′/3′ external transcribed spacer (ETS), 18S, 5.8S, 28S, internal transcribed spacer (ITS1/2), and transcription terminator factor 1 (TTF-1) site.
The upstream transcription enhancer elements (UTEEs), also known as the spacer promoter enhancer repeat, are another regulatory element located in the IGS [25]. This is the site of the formation of an enhancer boundary complex formed by CCCTC-binding factor (CTCF) and cohesion [24].
Upstream the enhancer boundary complex is flanked by nucleosomes, while downstream various components of the Pol I basal transcription apparatus were found, including Pol I, selectively factor -1 (SL-1), UBTF, RRN3, and TTF-1. The functional significance of these components is unclear as they are not involved in transcription. This is probably an artefact caused by the spatial proximity of the enhancer boundary complex and the core promoter, which leads to cross-linking of factors associated with the core promoter and the UTEE. It has been proposed that the enhancer boundary complex forms and serves as entry points for chromatin remodelling [24].
Traditionally, with exception of the IGS, which mainly contains repetitive sequences and transposable elements, the rRNA genes are believed to be highly conserved among species; however, increasing evidence suggest that not all rRNA genes are identical and instead exist in several variants [26,27,28]. The first report of a non-canonical rDNA repeat was by Caburet et al. in 2005, describing palindromic sequences arranged as mosaics with canonical repeats in human NORs [29].
The concept of heterogeneous rDNA repeats and copy number variation was further strengthened by two recent studies (Parks et al. and Wang and Lemos), which analysed whole genome sequencing data from the 1000 genome project [14] and different cancer types [13]. Parks and colleagues revealed that rDNA copy numbers varied greatly (about tenfold) between individuals within a human population and discovered pervasive inter- and intra-individual rDNA sequence variability. Interestingly, rRNA sequence variations are often associated with functionally important sites. For example, rRNA variations affecting inter-subunit bridge elements that establish the binding interface, linking the small and large ribosomal subunits impact on translation [14]. The same study described tissue-specific expression of rRNA variants in mice [14].
Analysis of over 700 tumours and corresponding normal tissues revealed a coupling of 5S rDNA array expansion with a loss of 45S repeats, which was most prevalent in TP53 mutant cancers. However, these variations were considered within the limits of natural variability and did not mediate an overall decrease in rDNA transcriptional output per cell. Two studies (Wang et al. and Ida et al.) hypothesized that loss of copies of 45S rDNA may be caused by replicative stress, as a result of rapid replication and high rDNA transcription rates affecting sister chromatid cohesion. They suggested that this loss would be beneficial for cancer cells as excessive 45S rDNA copy numbers are believed to promote genomic stability by facilitating recombinational repair [13,30]. The same study discussed the concept that loss of 45S copy numbers may be compensated for by epigenetic mechanisms controlling rDNA transcription. Thus, it is not surprising that loss of chromatin remodeller ATRX (α thalassemia/mental retardation X-linked), a member of the SWI/SNF family of helicase/ATPases, causes a substantial reduction in rDNA copy number [31].
3. Pol I Transcription Machinery
Three key “basal” Pol I transcription factors have thus far been identified; these are SL-1, Pol I associated regulatory factor RRN3 (also called TIFIA) and UBTF (also called UBF).
SL-1 is a complex of the TATA-binding protein (TBP) and 4 TATA-binding protein-associated factors (TAF), TAFI48 (TAF1A), TAFI63 (TAF1B), TAFI110 (TAF1C) and TAFI41 (TAF1D) [21,32]. SL-1 is responsible for promoter recognition [33] and together with UBTF, a high-mobility group (HMG) box protein (UBTF1 and UBTF2), drives the initial steps of pre-initiation complex (PIC) formation [34]. SL-1 binding to the CORE of the rDNA promoter is followed by binding of UBTF homodimers to both the CORE and upstream core element (UCE), which initiates rDNA promoter looping, bringing both regions into close proximity and thus stabilizing SL-1 [35,36,37]. How UBTF is recruited remains unclear; however, one study by van de Noebelen et al. suggested that CTCF may facilitate this process [38].
RRN3 has a unique HEAT repeat fold and regulatory serine phosphorylation sites [39]. RRN3 interacts with SL-1 to recruit the Pol I complex, the “core” Pol I subunits and auxiliary factors [40] to the promoter, thereby completing the formation of the PIC. After transcription initiation, RRN3 is released coinciding with the dissociation of the Pol I complex from promoter-bound initiation factors (promoter escape) [36,41,42,43].
UBTF not only acts as an archetypal transcription activator but also facilitates post-initiation events [36] via a variety of reported mechanisms. UBTF interacts with a heterodimer of two Pol I subunits, polymerase associated factor (PAF) 53 and CD3ε-associated signal transducer (CAST, also known as PAF49) to facilitate promoter escape [44]. In addition, UBTF regulates the elongation rate of Pol I by binding within the transcribed region of the rDNA where it forms so-called enhanceosomes, although this is still a matter of debate and awaits definitive structural analysis of UBTF on the rDNA [35]. In addition, UBTF binding affinity to the rDNA is regulated by differential phosphorylation [45,46,47,48].
More globally, UBTF1 has been reported to remodel chromatin, such as converting inactive rRNA genes into active chromatin by displacing linker histone 1 histones [24,49,50], as their binding is mutually exclusive, while UBTF2 exhibits no chromatin remodelling activity. Thus, UBTF1 is critical for the topological organization of the rDNA repeats. UBTF is also critical during mitosis as it remains constitutively bound to the rDNA pausing transcription; thus, it is thought to ‘bookmark’ active genes to facilitate re-initiation of transcription when cells enter the interphase [51]. This mitotic bookmarking enables UBTF to play a central role in the formation of NORs and maintaining secondary constrictions at active NORs [52,53]. Based on the UBTFs’ extensive binding across active genes and their absence in the IGS (except for the enhancer and promoter regions), it is considered a mark of euchromatic rDNA.
Around two decades ago, two distinct Pol I subcomplexes were identified initially in yeast and later in humans (Pol Iα and Pol Iβ) [40,41]. Both subcomplexes were catalytically active, but only one representing a small proportion of total cellular Pol I (2–10%) was competent to initiate transcription from the rDNA promoter. The competence to initiate transcription was determined by the presence of RRN3. The subcomplexes consist of 12–13 so-called “core” subunits but associate with a different set of auxiliary factors that further determine the functionality of these complexes. For example Pol Iα, which is the elongating form of Pol I, contains histone chaperone FACT (facilitates ATP-independent nucleosome remodelling) [54], whereas Pol Iβ, which is the initiating form of Pol I, is defined by the presence of RRN3, DNA topoisomerase (Top) IIα and casein kinase II [40,55,56,57,58,59]. As Pol I starts transcribing, the resultant nascent rRNA immediately associates with the pre-RNA processing machinery, tightly coupling rRNA synthesis and maturation [60]. In addition to the above “core” components of the Pol I transcription machinery, a multitude of other factors, such as nucleolin, nucleophosmin, Top I/II and chromatin-remodelling complexes [58,61,62], have been implicated in the regulation of Pol I loading and elongation. Finally, as mentioned above, termination of Pol I elongation and its dissociation is mediated by TTF-1 binding to the terminator elements [63].
4. Regulation of Ribosomal Gene Transcription
Pol I transcription is regulated in response to extracellular (e.g., environmental stimuli, stresses) or intracellular (e.g., cell cycle, cell growth) stimuli by a number of mutually non-exclusive mechanisms, including post-translational modification of the Pol I machinery, alterations in rDNA topology, changes in rDNA chromatin structure and through non-coding RNAs [64].
In response to environmental stimuli (e.g., growth factors) and various stresses (e.g., DNA damage), components of the Pol I transcription machinery are targeted by a number of signalling pathways, including PI3K-mTOR, RAF-MEK-ERK, AMPK and AKT signalling, which often converge, forming a complex signalling network [65,66,67,68,69,70,71,72,73]. Importantly, the severity and duration of cellular stresses (e.g., starvation, DNA damage, heat shock) induce differential activation of these pathways and alter the epigenetic landscape of the rDNA via chromatin modifiers and remodellers, leading to the changes in chromatin accessibility [74,75,76,77]. Apart from Pol I-specific transcription factors (TF), a range of other TF’s and oncogenes, previously only associated with Pol II-dependent transcription, have now been reported to also modulate rRNA synthesis. For example, acute myeloid leukemia (AML) 1-ETO, an AML-specific fusion-protein, has been reported to bind to human rRNA genes and promote Pol I transcription in malignant myeloid cells [78], while AML1 (Runx1) downregulates Pol I transcription [78].
In general, DNA topology is affected by transcription and replication. Top are a family of enzymes that release torsional stress at transcribed and replicated DNA loci. Two types of Top, Type I and Type II, relax supercoiled DNA by catalysing either single-strand or double-strand DNA breaks, facilitating DNA rotation or the passage of one DNA strand and re-ligation.
Experiments in yeast showed that Top I is involved in alleviation of the negative superhelical density formed behind elongating Pol I, whereas Top II is required for resolving positive supercoiling formed ahead of the transcription complex [79].
More recently, Denissov et al. revealed that regulatory elements, including the promoter, upstream region and terminator of actively transcribed genes, spatially interact throughout the cell cycle, forming so-called core–helix structures and Top I plays an essential role in maintaining this topology [80]. This rDNA loop formation brings the initiation and termination sites into close proximity, which was suggested as a means to facilitate the ‘recycling’ of Pol I complexes. A number of factors, including TTF-1 and c-Myc, have also been identified as being involved in this loop formation. Direct evidence of active rDNA looping was recently presented by Maiser and colleagues [81]. Ray et al. showed that, in human cells, Top IIα altered the rDNA topology at the rRNA core promoter and this was required for the assembly of functional PICs [58].
Non-coding RNAs were first described as regulators of rDNA transcription over a decade ago [82], specifically as being critical for rDNA silencing by facilitating the interaction between the nucleolar remodelling complex (NoRC) subunit TTF-1 interacting protein (Tip5) and TTF-1 [27,83]. These non-coding RNAs are proposed to promote heterochromatin formation at the rDNA and other chromosomal repeats [84,85] (for recent reviews, see [86,87]).
Interestingly, Abraham et al. proposed recently that Pol II-driven production of anti-sense transcripts originated from the rDNA IGS facilitated the formation of DNA–RNA hybrid structures, known as R-loops, at the boundaries of the IGS and coding rDNA regions. R-loops prevented Pol I-driven transcription of the IGS and also the production of sense intergenic noncoding RNAs (sincRNAs) that can negatively affect rRNA transcription [88]. These findings provide a potential direct mechanism that couples both Pol II and Pol I transcription activities.
5. rDNA Chromatin Dynamics
It is well established that chromatin undergoes extensive and dynamic remodelling in order to modulate gene transcription. While repositioning and remodelling of nucleosomes, plus modifications of histones at specific sites (e.g., promoters), are central to the control of gene transcription, earlier studies in yeast and Drosophila oocytes suggested that actively transcribed rRNA genes were nucleosome depleted [89,90]. However, this view was challenged by another chromatin study in yeast revealing that active rRNA genes were indeed associated with histones and nucleosomes [91]. Currently, there is no consensus as to whether in higher eukaryotes actively transcribed rDNA are associated with functional nucleosomes or core histones, such as those typically located on Pol II-transcribed genes. A number of studies have demonstrated that histones H3 and H4 are associated with transcribed rDNA in human cells [74,92], while other studies disagree [24,87].
Integrative genomic analysis of human and mouse embryonic stem cells (ESCs) revealed similarities in the enrichment of euchromatic and heterochromatic histone marks ~2 kb upstream of the rDNA core promoter, while the other regions within the rDNA were markedly different [93,94]. A recent study by Herdman and colleagues reported active histone marks (H3K4me2/3, H2A.Z/ac, H3K9ac, H3K27ac and H3K36me3) exclusively in the enhancer region but not within the transcribed region or the IGS upstream of the enhancer boundary complex [24]. Interestingly, in nutrient-starved cells, histone H3 was found associated with the transcribed regions of active rDNA repeats, but this was rapidly removed when the cells recovered from starvation [74], highlighting the dynamic nature of rDNA chromatin.
A landmark study in 2008 presented evidence that, in fact, the rDNA repeats exist in three distinct chromatin configurations: (i) transcriptionally active rDNA repeats that are hypo-CpG methylated at the promoter-specific CpG, enriched for euchromatin histone marks and bound by UBTF; (ii) pseudo-silent/poised rDNA repeats that are hypo-CpG methylated at the promoter, bear repressive histone modifications, but are not bound by UBTF, and thus exist in a closed chromatin conformation; and (iii) silent rDNA repeats that are promoter hyper-CpG methylated and associated with heterochromatic histone marks adopting a highly compact chromatin state [50]. The silencing of the rDNA repeats is promoted by NoRC, a complex which recruits histone and DNA modifiers such as histone deacetylases 1 (HDAC1) and DNA methyltransferase 1/3a (DNMT1/3a) to the rDNA [76].
In contrast to the promoter-associated CpG methylation, the presence of CpG methylation within the rDNA coding region and its association with active transcription is still a matter of debate and complicated by their repetitive multi-copy nature. A recent study by Wang and Lemos reported that the rDNA CpG hypermethylation strongly correlated with aging, thus serving as an evolutionarily conserved biological clock [95]. However, no link between age-mediated methylation and the level of rRNA transcription has been reported.
The transcriptional pseudo-silent/poised rDNA chromatin conformation is established by nucleosome remodelling by deacetylation complexes (e.g., NuRD [96] and eNoSc [75]). While these poised rDNA promoters are unmethylated and have nucleosomes positioned to prevent transcriptional initiation, they are often characterized by either bivalent histone modifications (e.g., H3K4me3 and H3K27me3) or fully repressive histone marks (e.g., H3K4me1/2 and H3K9me3). Re-activation of transcription in this case will depend on the re-positioning of a nucleosome at the rDNA promoter by the DNA-dependent ATPase Cockayne syndrome protein B (CSB) [96] and histone modifications by the coordinated actions of histone methyltransferases (e.g., MLL1–2 [97]), histone demethylases (e.g., PHF8 [98], KDM4A [74] and G9a [99]) and histone acetyltransferases (e.g., PCAF [100]). It is feasible to propose that these enzymes are not acting alone but are in fact part of a large activating complex. This idea is supported by data showing that many chromatin modifiers involved in the activation of rDNA transcription interact with the scaffold protein WD repeat-containing protein (WDR)5 [101,102].
Despite acknowledged species-specific differences in nucleosome occupancy at actively transcribed ribosomal genes, this remains a “hot-topic” of discussion. To add to the epigenetic complexity of rDNA chromatin, a number of non-canonical histone variants have been identified that bind to the rDNA. For example, histone H3.3, a variant previously described to be involved in transcriptional activation as well as gene silencing, was recently demonstrated to bind to the rDNA [31]. Other histone variants, e.g., H2AZ, was located on the IGS region of the rDNA and incorporated into the rDNA under high glucose conditions [103,104]. Interestingly, phosphorylated H1.2 and H1.4 variants of histone H1, which are commonly associated with inactive rDNA chromatin, were found to promote transcription of the rDNA genes [103,105].
In addition to histones and Pol I-specific TFs, there are ubiquitous DNA-binding proteins, such as CTCF, and structural maintenance of chromosome (SMC) complexes, such as cohesin and condensin [38,106,107], which can bind and modulate the epigenetic state of the rDNA genes. CTCF binds upstream of the rDNA spacer promoter and interacts with UBTF and other Pol I complex components, suggesting it is a regulator of Pol I transcription and rDNA chromatin. This was further supported by the finding that CTCF depletion reduced UBTF and Pol I binding near the spacer promoter [38]. Although these proteins are not considered components of the core transcription machinery, they are integral to the structure of rDNA chromatin [24,87,108].
Overall, regulation of rDNA chromatin structure plays a pivotal role in maintaining the balance between actively transcribed, pseudo-silenced and silenced rDNA repeats, and thus is crucial for controlled responses to, e.g., stress, development, aging and genomic instability. Despite the identification of a number of key players, our understanding of these complex mechanisms, and in particular the crosstalk between them, is still limited, and the focus of ongoing research.
6. Role of the Nucleolus in Spatial Genome Organization and Pol II Transcription
The overall three-dimensional (3D) organization of the genome highly depends on the formation of chromatin contacts within and/or between each chromosome and the nuclear domain. These types of interactions include: (i) chromatin loop formation that bypass long genomic distances and connect distant genomic regions, such as enhancers and promoters; (ii) the formation of topologically associated domains (TADs) that are local-interacting DNA neighbourhoods; and (iii) regions where genomic loci interaction is driven by their transcriptional activity.
Several studies have demonstrated a direct role for the nucleolus in genome organization and as a global modulator of all transcription (Pol I, II and III) [9,10,11,109,110,111]. The nucleolar periphery contains satellite DNA repeats that form a perinucleolar heterochromatic dense shell. Specific genomic regions located in the perinucleolar region are called nucleolus-associated domains (NADs) (for a recent review, see [110]), whereas those associated with nuclear lamina are called LADs; however, in humans, a proportion of NADs and LADs overlap (Figure 2). NADs were first described in 2010 by two groups, van Koningsbruggen et al. and Nemeth et al., who demonstrated that specific genomic sequences interact with the nucleolus (around 4% of the genome) [9,11]. Using complementary approaches of fluorescence in situ hybridization (FISH) and DNA sequencing, they described the NAD-interacting genomic domains as being generally located in gene-poor regions but did identify a number of specific gene families, including zinc-finger, olfactory receptor and defensin genes, as well as satellite pericentromeric and centromeric repetitive sequences, regions of the inactive X-chromosome (Xi), as being enriched with NADs. Further, some tissue-specific expressed gene clusters were reported as associated with NADs, including two immunoglobulin clusters and T-cell receptor genes [9,11]. Analysis of RNA coding genes revealed an enrichment of Pol III-dependent 5S and transfer RNA genes in NADs, suggesting that their spatial organization within the nucleus may play a key role in their transcriptional regulation. This idea was further supported by the finding that NADs are enriched in repressive histone marks, such as H3K27me3, H3K9me3 and H4K20me3, while active marks are excluded, which correlated with a decrease in global gene expression of NAD-associated loci [9]. In fact, NADs are nuclear territories with their proposed primary function being the maintenance of heterochromatin at interacting regions. For example, the Xi continuously revisits the nucleolus during cell cycle progression through S phase to maintain its heterochromatic state [112].
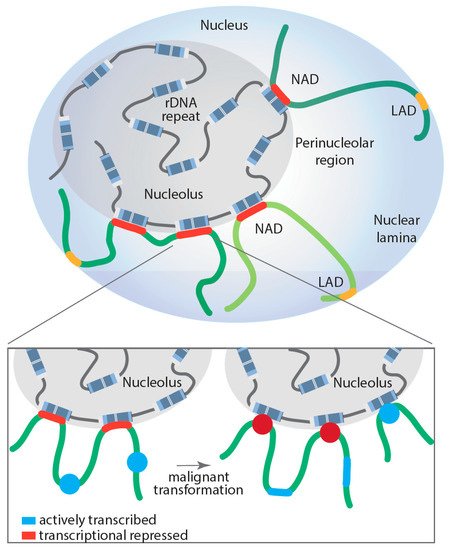
Figure 2. Proposed model of dynamically regulated long-range rDNA–NAD interactions during malignant transformation and their impact on Pol II-dependent transcription at associated loci. Lamina-associated domain (LAD); nucleolus associated domain (NAD); red: transcriptionally repressed Pol II-dependent genes; blue: actively transcribed Pol II-dependent genes.
However, NADs are not solely enriched on transcriptionally silenced genes; a number of highly transcribed Pol III-dependent RNA genes are also NAD associated. The dynamic regulation of the NAD interactions with genomic loci still requires intensive study. Comparative Hi-C analysis of NAD interactions in human embryonic fibroblasts revealed that surprisingly most of these interactions remained unchanged when comparing proliferating and senescent cells, with the exception of specific satellite sequence clusters that segregate from the nucleoli after cells underwent replicative senescence [113]. To shed more light onto the dynamic nature of NAD interactions and whether those interactions may contribute to disease development, Diesch and colleagues mapped genomic loci interacting with the nucleolus during malignant transformation [10]. Remarkably, the study demonstrated that Myc-driven malignant transformation of B-cells is associated with a significant increase and reorganization of rDNA-NAD contacts due to activation of previously silent rDNA genes (rDNA class switching). This spatial rearrangement correlated with gene expression changes at associated genomic loci, impacting the Pol II-transcribed gene ontologies, including B-cell differentiation, cell growth and metabolism, changes that contribute to malignant cell fitness [10]. Moreover, these studies support, for the first time, a model where structural changes in rDNA chromatin and subsequent rDNA-NAD reorganization promote gene expression changes at associated loci, which influence clonal selection of a malignant cell population (Figure 2).
This entry is adapted from the peer-reviewed paper 10.3390/genes12050763
This entry is offline, you can click here to edit this entry!