Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Neurosciences
Autophagy plays critical roles in development, maintenance and survival of distinct cell populations including neurons.
- ageing
- autophagy
- neurodegeneration
1. Mechanisms of Autophagy
Autophagy is an evolutionarily conserved process enabling cells to regulate a plethora of catabolic needs. Under physiological conditions, autophagy serves as a quality control mechanism in order to maintain cell homeostasis. Importantly, in response to extracellular or intracellular stress, autophagy is employed to degrade and recycle macromolecules such as misfolded or aggregated proteins as well as to eliminate dysfunctional organelles or invading pathogens [1,2,3]. Macroautophagy, microautophagy and CMA (chaperone-mediated autophagy) are the three morphologically and mechanistically distinct types of autophagy. However, all three converge towards the delivery of cargo to the lysosome for degradation.
In microautophagy, cytoplasmic components enter the lysosome through a direct invagination of the lysosomal limiting membrane. Membrane fission results to the release of a microautophagic body into the organelle lumen for degradation [4]. CMA involves the selective recognition of a substrate based on the KFERQ-like motif by a cytosolic chaperone, the HSC70 (heat shock cognate 71 kDa protein) and cochaperones [5]. Subsequently, unfolding of the substrate and translocation via the receptor LAMP2A (lysosome-associated membrane protein type 2A) leads to the degradation of the substrate.
Macroautophagy (hereafter referred to as autophagy), is a process that enables cells to recycle and degrade intracellular contents. Hallmark of autophagy is the de novo formation of a double-membrane vesicle, termed the autophagosome, which sequesters and engulfs cytoplasmic material (Figure 1A). Subsequently, autophagosome matures and fuses with a lysosome to form an autolysosome, leading to the exposure of the autophagic cargo to the lysosomal hydrolases for digestion [6,7,8]. Autophagosome formation is driven by the orchestrated action of approximately 20 ATG (autophagy-related) proteins, whose role is essential for autophagic delivery of cargo to the lysosome or vacuole in yeast (Figure 1B,C) [9,10,11,12,13]. Nutrient and energy sensing pathway hubs, including mTOR (mechanistic target of rapamycin) and AMPK (AMP-activated protein kinase), converge to the ULK1 (Unc-51-like kinase 1) complex [ULK1, ATG13, FIP200/RB1CC1 (200 kDa FAK family kinase-interacting protein/RB1-inducible coiled-coil protein 1) and ATG101] for autophagy initiation (Figure 1B). ULK1 complex triggers the nucleation of an autophagosome membrane precursor, termed isolation membrane or phagophore by phosphorylating members of the PI3KC3 (class III phosphatidylinositol-3 kinase) complex I [VPS34 (vacuolar protein sorting 34), Beclin 1, ATG14, AMBRA1 (activating molecule in Beclin 1-regulated autophagy protein 1) and general vesicular transport factor (p115)]. PI3KC3 complex I induces the local production of PI3P (phosphatidylinositol-3-phosphate) at a characteristic endoplasmic reticulum structure called the omegasome. Two PI3P effector proteins the WIPI2 (WD repeat domain, phosphoinositide interacting 2) and the DFCP1 (zinc-finger double FYVE-containing protein 1) are recruited to the omegasome via interaction with their PI3P-binding domains. Locally accumulated WIPI2 directly binds and recruits ATG12 (ATG12-ATG5-ATG16L1) complex and catalyzes the conjugation of the C-terminal glycine of ATG8 family proteins, [including LC3 (microtubule-associated protein light chain 3) and GABARAP (γ-aminobutyric acid receptor-associated protein)] to the phagophore-resident lipid PE (phosphatidylethanolamine) (Figure 1C). The lipidated, thus membrane-bound LC3 (called LC3-II), characteristic signature of autophagic membranes, is required for elongation and closure of the phagophore. ATG4, the sole ATG protease, processes nascent ATG8 to expose the C-terminal glycine residue [14]. ATG8 protein family members interact with autophagy receptor proteins or SARs (selective autophagy receptors) bearing LIRs (LC3-interacting regions) in order to sequester the cytoplasmic material destined for lysosomal degradation [15]. The endoplasmic reticulum (ER) and the majority of cellular membranes have been proposed to contribute membrane material for the elongation of the autophagosomal membrane [16,17]. ATG2 mediates phospholipid transfer from ER or other donor membrane sources to an ATG9-positive vesicle/phagophore. ATG9, the sole transmembrane ATG protein possesses membrane-bending properties. It facilitates autophagosomal membrane expansion acting as a scramblase. Specifically, ATG9 transports phospholipids from the cytoplasmic to the luminal leaflet of the isolation membrane [18,19,20,21,22]. ESCRT (endosomal sorting complexes required for transport) machinery facilitates phagophore closure, a process involving membrane fission of the inner and outer membrane at the phagophore edge [23,24]. After phagophore closure, the complete autophagosome undergoes maturation by dissociation or removal of most ATG proteins, including ATG8 by ATG4 protease, as well as PI3P by MTMR (myotubularin-related) phospholipid phosphatase from the autophagosome surface [25]. ATG8 family members, tethering factors, Rab (Ras-associated binding) GTPases, and SNARE (soluble N-ethylmaleimide-sensitive factor-attachment protein receptor) proteins act in concert to mediate fusion of autophagosomes with lysosomes [26]. Finally, the lysosomal acidic hydrolases digest the sequestered autophagic cargo and salvaged building blocks including amino acids, fatty acids, nucleotides and sugars are recycled back to the cytoplasm in order to be used again by the cell.
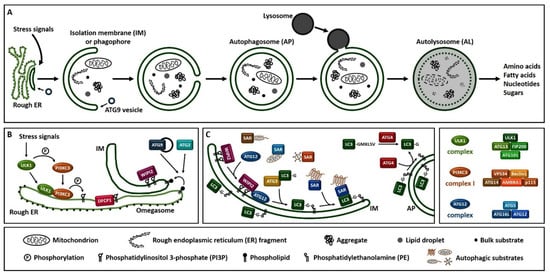
Figure 1. Overview of the autophagy process in human. (A). Schematic representation of autophagosome formation and cargo degradation. Complete autophagosomes may fuse with endosomes to form amphisomes, which further fuse with lysosomes. (B). Omegasome and isolation membrane generation. (C). Isolation membrane expansion, LC3 processing and autophagic substrate sequestration. Additional membrane sources may contribute to isolation membrane formation and expansion. SAR: selective autophagy receptor, LC3-GKLSV: pro-LC3, LC3-G: LC3-I, LC3-G-PE: LC3-II.
Autophagy is considered to have primarily cytoprotective and pro-survival functions. Therefore, as a critical cellular process it needs to be tightly regulated in order to respond and adapt appropriately to various cellular stress signals and insults. Several mutations in ATG genes have been identified and associated with human genetic disorders, developmental abnormalities, immune diseases, cancer and neurodegeneration, demonstrating a role for autophagy in animal health [27,28].
2. Neuronal Autophagy
As anticipated, autophagy is indispensable for neuronal survival and maintenance of neuron homeostasis. Genetic studies have demonstrated that conditional ablation of the core autophagy genes atg5 and atg7 in the nervous system of mice leads to accumulation of ubiquitin-containing inclusion bodies disrupting neuronal function and causing progressive neurodegeneration [29,30]. Autophagy in hypothalamic neurons is required specifically for the regulation of nutrient uptake, energy balance, obesity as well as ageing [31,32,33,34]. Hippocampal neurons of fragile X syndrome mice model exhibit reduced levels of autophagy. Activation of autophagy partially rescues the aberrant dendritic spine morphology, mGluR-LTD (metabotropic glutamate receptor-dependent long-term depression) and cognition in those mice [35]. Knockout studies have demonstrated an essential role for autophagy in embryonic neurogenesis and mutations in autophagy genes lead to developmental delay, cognitive decline, and functional deficits in childhood [36,37,38,39,40,41,42,43,44,45,46,47,48,49]. In addition, autophagy plays a role in the differentiation of adult NSCs (neuronal stem cells) [50,51,52,53,54,55,56,57]. Furthermore, specific processes involved in neuronal plasticity such as axonal growth, synaptic assembly, and dendritic spine formation and pruning are linked to autophagic activity and accompanied by cognitive deficits, anxiety-like behaviors, autism-like behaviors and memory deficits [35,58,59,60,61,62,63,64,65]. Given the contribution of autophagy in neurogenesis and neuronal plasticity, it is apparent that pharmacological or genetic induction of autophagy might be a means of treatment and therapy for disorders like depression, bipolar disorder, and schizophrenia [66,67,68,69,70,71,72,73,74,75,76,77]. In contrast, the lysosomal acidification inhibitor bafilomycin A1, which blocks autophagosome-lysosome fusion and therefore autophagic flux, has antidepressant effects in rats exposed to chronic unpredictable mild stress [78]. In that case, autophagy inhibition seems to be advantageous.
Neurons possess unique structural and functional characteristics. Cellular processes have to be tightly and differentially regulated in a spatiotemporal manner. For example, neuronal soma is often located far away from the synapses. Therefore, specialized processes restricted to the microenvironment of the synapse ensure proper synaptic transmission. Synaptic activity has been linked to autophagy in molecular and vesicular level [61,79,80,81,82,83,84,85]. Constitutive de novo autophagosome biogenesis occurs at the axon terminal and upon completion, autophagosomes fuse with late endosomes and/or lysosomes. In distal axons of primary DRG (dorsal root ganglion) neurons, autophagosomes seem to be generated at DFCP1-positive subdomains of the endoplasmic reticulum, distinct from ER exit sites [86]. Moreover, plasma- or mitochondrial-derived membranes were not incorporated into nascent autophagosomes. A minor population appears to arise from pre-existing autophagosome rings. The authors suggest that autophagosome rings may sometimes nucleate other smaller autophagic structures. Subsequently, autophagosomes are transported along the axonal microtubules toward the soma, in a dynein-dependent manner [87,88,89,90,91,92]. In transit, they fuse with additional lysosomes, arriving at the soma as fully competent degradative organelles. In general, autophagosome biogenesis events are enriched distally. However, autophagosomes form infrequently in dendrites, the soma, or midaxon [86].
Presynaptic autophagosomes engulf synaptic vesicles and therefore autophagy regulates neurotransmission by controlling the pool of the synaptic vesicles and neurotransmitter release [60,79]. Pharmacological activation of autophagy reduces the number of synaptic vesicles. Loss of autophagy increases evoked dopamine release in mice. BDNF (brain-derived neurotrophic factor) stimulates retrograde motility of autophagic compartments positive for the receptor of BDNF, TrkB (tropomyosin receptor kinase B) [93]. The phosphorylated endocytic adaptor endophillin A is enriched at the presynaptic terminals and promotes autophagy by generating highly curved membranes, where core autophagy proteins are recruited to form autophagosomes [82,83]. The lipid phosphatase synaptojanin 1, implicated in synaptic vesicle trafficking, is also required for autophagosome biogenesis at presynaptic terminals [61]. The scaffolding protein bassoon, localized to the presynaptic nerve terminals as well, sequesters ATG5 and inhibits presynaptic autophagy [60]. Mitophagy targets ubiquitinated mitochondria in synapses; thereby it may regulate local energy supply or calcium buffering capacity [94].
Predominant destination for autolysosome cargo degradation is the cell body, where lysosomes with high proteolytic activity reside [95,96]. Inhibition of lysosomal activity leads to accumulation of autophagosomes specifically within the soma, and not in the axon or dendrites [97]. Sequestration of mitochondria fragments have been captured into a subpopulation of autophagosomes [98]. Interestingly, an alternative mechanism for eliminating protein aggregates and mitochondria when autophagy is compromised is the secretion of large vesicles called exophers by several neuronal cell types, including dopaminergic and sensory neurons [99,100]. Extruded exophers are engulfed by neighboring cells for lysosomal degradation or released into the extracellular milieu. These finding suggest that neuronal cells do not necessarily degrade their own aggregates or damaged organelles, introducing a possible mechanism for the prion-like propagation phenomenon. From a pharmacological point of view, on one hand, induction of the release of toxic cargo outside of the cells will probably relieve the neurons from toxic loads but increase the propagation of prion-like pathologies. On the other hand, inhibition of the jettison of aggregates or damaged organelles, will probably prevent the pathology from spreading but will increase the toxic load into the neurons with possible detrimental consequences.
Postsynaptic autophagy plays a role in synaptic plasticity and synaptic strength, which are fundamental processes underling learning and memory [101,102,103,104]. Deletion of WDR45 (WD repeat domain 45) protein at the central nervous system leads to deficits in learning and memory in mice [105]. In LTP (long-term potentiation), low-dose activation of NMDARs (N-methyl-D-aspartate receptors) leads to an increase in AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) internalization [106]. Conversely, in LTD (long-term depression), activation of NMDARs results in AMPARs degradation in hippocampal neurons. BDNF suppresses autophagy in cortical and hippocampal neurons, facilitating LTP and the persistence of memories in mice through stabilization of the postsynaptic scaffold proteins PICK1 (protein interacting with C kinase 1), PSD-95 (postsynaptic density protein 95) and SHANK3 (SH3 and multiple ankyrin repeat domains 3) [107]. Upon denervation of neuromuscular junctions, GABAARs (gamma-aminobutyric acid type A receptors) are selectively sorted via endocytosis from the postsynaptic membrane of muscle cells to autophagosomes, whereas acetylcholine receptors in the same cells do not localize in autophagosomes [108].
Interestingly, non-canonical roles of autophagic machinery and ATG proteins have been linked to neuronal biology. ATG16L localizes on hormone-containing dense-core vesicles through interaction with RAB33 and participates in hormone secretion from neuroendocrine PC12 cells independently of its role in autophagy through RAB33 [109,110]. LANDO (LC3-associated endocytosis) regulates amyloid-β clearance in microglia [111]. The inflammatory cytokine IL-1β (interleukin-1β) is secreted via an unconventional autophagy-mediated secretory pathway in human neutrophils [112].
3. Autophagy in Ageing
Ageing, the time-dependent decline in cellular, tissue and organismal functions occurs in all metazoan organisms. Genetics play fundamental role in controlling ageing [114]. Numerous mutations that either extend lifespan or accelerate age-related decline have been characterized. Studies in those mutated genes revealed specific signaling pathways and cellular machineries conserved among species that regulate ageing. Accumulating evidence supports autophagy as a critical regulator of lifespan [115,116]. Caloric restriction and mTOR inhibition, two interventions known to extend lifespan, induce autophagy [116]. Autophagic activity and efficiency declines with age in diverse organisms. From C. elegans and rats to primary human cells, ageing reduces the capacity of lysosomal proteolysis compared to their younger counterparts [117,118]. Reduced expression of several ATG genes upon ageing is documented in several organisms including Drosophila and rodents [119,120,121]. In Drosophila, loss-of-function mutations of Atg7 and Atg8 genes reduce lifespan, increase sensitivity to stress and promote neuronal accumulation of ubiquitin-positive aggregates [119,122]. Mice exhibit an age-dependent decrease in autophagosome numbers [123]. In addition, genes important for autophagosome-lysosome fusion such as LAMP2, show reduced expression upon ageing [124]. In human brain, ATG5, ATG7 and BECN1 are down-regulated during normal ageing [125]. Individuals with age-associated neurodegeneration diseases possess autophagy gene polymorphisms and exhibit reduced autophagy [126,127,128,129]. Since impairment of autophagy predisposes organisms to age-related diseases, such as neurodegeneration, genetic or pharmacological restoration of autophagy may be utilized for improving ageing-related diseases [115].
Oxidative stress, DNA damage, telomere shortening and inflammation play prominent causative role in ageing [130,131,132,133]. These factors can compromise cellular proteostatic mechanisms such as autophagy and contribute to the development or progression of age-related neurodegenerative disorders, such as Alzheimer’s, Huntington’s or Parkinson’s disease, and amyotrophic lateral sclerosis [134,135,136,137,138].
Research at the interface of autophagy and ageing highlights interesting and elegant aspects of this interplay [139]. Lifespan extension can be achieved with tissue-specific overexpression of a single atg gene, revealing the minimum intervention with sufficient effect in organismal level. Furthermore, autophagy stimulation in a select tissue can have systemic effects and influence ageing in a cell-non-autonomous manner. Finally, targeting and clearance of specific dysfunctional components via selective types of autophagy may be sufficient for longevity, highlighting the importance of selectivity and avoidance of excessive off-target energy-consuming autophagic activity. Focusing on research in the nematode C. elegans, we summarize recent advances in our understanding of the role of general and selective neuronal autophagy modulation in neuroprotection as well lifespan and healthspan regulation.
This entry is adapted from the peer-reviewed paper 10.3390/cells10030694
This entry is offline, you can click here to edit this entry!