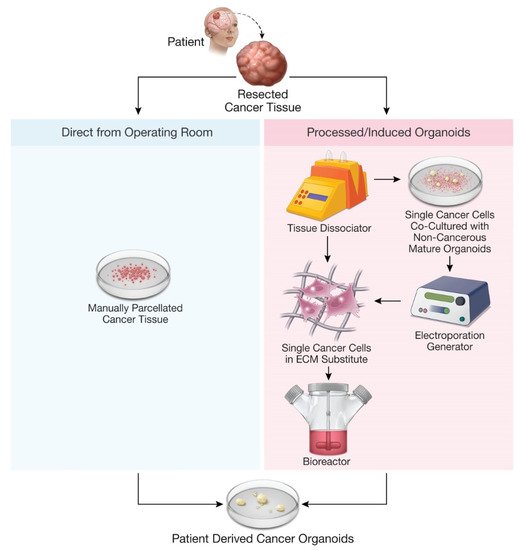
The emergence of three-dimensional human organoids has opened the door for the development of patient-derived cancer organoid (PDO) models, which closely recapitulate parental tumor tissue.
1. Introduction
The recent development of organoids, defined as in vitro three-dimensional models of human tissues, has allowed for more accurate recapitulation of parent organs [6,7,8,9,10,11,12,13,14,15,16,17]. The available literature describing patient-derived cancer organoids suggests that organoid cancer models consistently reflect in vivo tumor genotypes, phenotypes, intertumor heterogeneity, and cell–cell and cell–stroma interactions and are less resource and time intensive compared to existing mouse models [23,24,25,26,27,28,29,30,31,32]. Organoid cancer models may mitigate challenges presented by existing cancer models and augment the implementation of precision oncology by providing personalized platforms to study each patient’s tumor.
2. History of Organoid Development
Historically, the term “organoid” referred to any in vitro culture whose characteristics resembled a primary organ. However, within the past 15 years, an organoid has been redefined as a self-organizing three-dimensional structure made from murine or patient-derived stem cells or primary tissue [
32,
33,
34,
35]. Advances in stem cell science have facilitated the development of organoid cultures, particularly through the isolation of embryonic and pluripotent stem cells, which provided the tinder for the creation of early original organoid models [
36,
37,
38,
39]. Sato et al. (2009) were the first to describe organoids using intestinal stem cells for the induction and perpetuation of a human intestinal organoid [
36]. By using Lgr5
+ intestinal stem cells embedded in an extracellular matrix substitute (Matrigel) on media enriched with r-spondin 1, Noggin, and epidermal growth factor, the group was able to create crypt–villus organoid structures that retained the cellular organization and distribution of newly isolated small-intestinal crypts for over 8 months [
36]. Since 2009, organoid models have been used to study the development of several tissues, including the brain, retina, lung, thyroid, blood vessels, small intestine, liver, mesodermal kidney, taste buds, pancreas, and ovaries [
32,
37,
39,
40,
41,
42,
43,
44]. Notably, Lancaster et al. (2014) were the first to describe cerebral organoid models derived from human pluripotent stem cells that could produce a functional cortex, retina, and choroid plexus with discrete brain regions in a 1–2-month time frame [
37,
45]. Following this, fused dorsal and ventral cerebral organoids demonstrated directional migration and connectivity of GABAergic neurons [
44]. Together, the cerebral organoids created by these groups not only offered distinct advantages over standard neuronal culture models but also showed that creation of non-epithelial organoid models was possible and could recapitulate in vivo tissue architecture.
Several organoid models have now been described with protocols that use induced pluripotent, embryonic, or adult stem cells [
26,
30,
36,
37,
45,
46,
47,
48,
49,
50,
51,
52]. The organoid creation protocols for induced pluripotent stem cells and embryonic stem cells are similar, wherein isolated stem cells are exposed to tissue-specific growth factors to create desired embryoid body-like aggregates [
34]. The aggregates are then embedded in a scaffold that acts as the extracellular matrix, most commonly Matrigel or collagen, as this allows the stem cell aggregates to develop the three-dimensional architecture that is critical for the development of tissue polarity from cell–cell and cell–stroma interactions [
34,
53]. The embedded embryoid bodies are then cultured in media containing various tissue-specific growth factors to produce mature organoids [
34]. Alternatively, adult stem cells can also be used to create organoid models, but unlike induced pluripotent and embryonic stem cells, adult stem cells do not require genetic transduction [
32,
38,
54,
55]. Therefore, organoids created from adult stem cells can be immediately embedded in an extracellular matrix substitute and cultured in media enhanced with growth factors [
32]. The use of stem cells to create organoids, however, is limiting, as many require genetic engineering to produce models of pathologic tissue, which may not recapitulate in vivo disease, prompting groups to create tissue-derived organoid models of disease [
56,
57].
Organoids have now been developed to model pathologies, including genetic diseases, infectious diseases, parasitic infections, and cancer [
8,
58]. A primary focus of organoid disease models has been in modeling of cancer, which has resulted in several cancer organoid models derived from epithelial tumors—prostate, liver, pancreas, ovary, bladder, lung, gastrointestinal, and breast, among others () [
23,
24,
25,
26,
27,
28,
29,
30,
31,
59]. Some of the initial cancer organoid models used genetic modification of stem cells to produce cancer-like organoids, but recent work has focused on developing patient-derived cancer organoid (PDO) models from fresh cancer tissue [
26,
47,
49,
56,
57,
60]. The production of PDOs involves direct coordination between physicians performing tumor biopsy and laboratory personnel in order to create and propagate patient-specific cancer organoids, which may better represent the parent tumor compared to stem-cell-derived cancer organoids [
23,
24,
25,
26,
27,
28,
29,
30,
31,
59,
61,
62]. Biopsied tumor samples used for organoid production are optimally collected from specimens that are highly cellular, not cauterized, necrotic, or marginal samples with surrounding normal tissue as these factors are critical to organoid production success rates and clinical relevance [
61,
62]. One method to produce PDOs from patient tissue samples entails enzymatic digestion to isolate single cells, especially cancer stem cells, which are then propagated like adult stem cells [
35]. While the use of cancer stem cells is one way to create PDOs, this method has been criticized for limiting the clonal heterogeneity of resultant PDOs; hence, most PDO protocols now describe the use of fresh tissue samples that are parcellated but not enzymatically digested to single cells [
24,
26,
30,
31,
32]. After being parcellated, the tissue samples are cultured in specific media with growth supplements, often embedded in Matrigel, and incubated for weeks to months to produce mature PDOs () [
24,
26,
30,
31,
32]. For example, pancreatic ductal adenocarcinoma, which remains treatment refractory despite advancements in multimodality therapy, has been modeled with PDOs to test potential treatments [
24]. Breast cancer has also been modeled with PDOs, with a landmark study by Sachs et al. (2018) establishing culture conditions necessary for mammary epithelial organoid creation and propagation [
28].
Figure 1. Methods for production of patient-derived organoids.
3. Current Models of Patient-Derived Cancer Glioma Organoids
Several glioma organoid models have been described in the literature (). The methods used to create glioma organoids include isolation and culture of patient-derived stem cells, CRISPR/Cas9-mediated mutagenesis of non-cancer cerebral organoids, co-culture of GBM spheroids with normal cerebral organoids, and direct culture of patient-derived glioblastoma samples [
56,
57,
62,
82,
83]. Hubert and colleagues (2016) first described a GBM organoid model produced from fresh tumor stem cells that recapitulated hallmark features such as hypoxia gradients, selective radiosensitivity, and invasiveness when orthotopically xenografted [
57]. The glioblastoma-like organoids created from stem cells with or without directional mutagenesis were also capable of recapitulating some aspects of gene expression and phenotypic behavior of invasive brain tumors [
56,
68,
83,
84]. Further, co-culture techniques of glioma stem cells (GSCs) and spheroids with normal cerebral organoids offer significant advantages over xenograft models [
82,
83,
85]. Single-cell RNA sequencing of co-culture of patient-derived GSCs with cerebral organoids demonstrated more accurate representation of the parent tumor compared to glioma spheres, GSC-derived organoids, and xenograft models, indicating maintenance of the tumor microenvironment [
85]. However, these stem-cell-based models are limited by the need for exogenous growth factors (which may lead to population drift), reliance on mutagenesis to produce tumor-like phenotypes, use of Matrigel rather than the native extracellular matrix, and relatively slow growth rates [
56,
57,
68,
83,
84,
85].
Table 2. Glioma organoid models.
| Cancer Type |
Source of Organoids |
Culture Technique |
Endpoint of Study |
Resemblance to Parent Tumor |
References |
| Glioblastoma |
Patient-derived glioma stem cells |
1. Co-culture of GSCs and iPSCs
2. Supplementing GSCs with normal cerebral organoids
3. Fusion of GSC spheres with normal brain organoids |
Three techniques to model GSC invasion in normal brain organoids and creation of GBM organoids |
No direct comparison to primary tissue could be made. |
Goranci-Buzhala et al. (2020) [83] |
| Patient-derived glioblastoma and non-glioblastoma stem cells |
Matrigel + culture shaking in NBM complete media |
Spatial distribution of GBM replicated in organoids derived from glioblastoma and non-glioblastoma stem cells |
Orthotopically implanted PDOs were diffuse and infiltrative, histologically resembling the parent tumor. |
Hubert et al. (2016) [57] |
| Human embryonic stem cells |
Normal cerebral organoids co-cultured with patient-derived tumor cells or oncogene introduction through electroporation |
Normal human-derived cerebral organoids a vector for glioblastoma organoid modeling |
Engineered PDOs displayed a mesenchymal phenotype consistent with patient-derived GBMs on transcriptomic analysis. |
Ogawa et al. (2018) [56] |
| Human glioblastoma tissue |
Patient tissue parcellated and cultured without an extracellular matrix |
Glioblastoma organoid protocol development from primary tissue samples with minimum processing |
PDOs had cellular and nuclear atypia, abundant mitotic figures, and pleomorphic nuclei consistent with high-grade parent tumors on histologic analysis. |
Jacob et al. (2020) [62] |
| Glioblastoma spheroids infiltrating cerebral organoids |
Co-culture of mouse early (6-day-old) cerebral organoids with glioblastoma spheroids created from glioblastoma stem cell culture |
Demonstration of hybrid glioblastoma organoid modeling |
Co-cultured organoids displayed core infiltration and expression of GBM stem cell markers NESTIN and SOX2. |
Da Silva et al. (2018) [82] |
| Patient-derived glioma stem cells |
Co-culture of cerebral organoids with transduced GSCs on NBM |
Single-cell RNA sequencing comparison of four patient-derived glioblastoma models |
Organoids displayed microscopic invasion and single-cell heterogeneity consistent with parent tumors. |
Pine et al. (2020) [85] |
| Central nervous system primitive neuroectodermal-like and glioblastoma-like tumor |
Human embryonic stem cells |
Neoplastic cerebral organoids (Neo-COR); combination of plasmids introduced into normal cerebral organoids through electroporation before te organoids embedded in Matrigel |
Demonstration of brain tumorigenesis through introduction of oncogenic mutations in normal cerebral organoids through transposon and CRISPR/Cas9 mutagenesis |
GBM-like Neo-COR displayed upregulation of GBM genes on transcriptomic analysis. |
Bian et al. (2018) [68] |
Abbreviations: GBM, glioblastoma; GSCs, glioma stem-like cells; NBM, neurobasal medium.
Jacob et al. (2020) recently described successful GBM organoid production and biobanking from minimally processed fresh GBM samples [
61,
62]. Their model represents a significant advance from prior models due to several methodologic advantages and biological similarities to human tumors [
62]. These GBM organoids were created with fresh tissue samples taken from the operating room in a medium formulated for neural tissue, clipped to appropriate size, and washed with minimal processing [
61]. The pieces were subsequently cultured in basal media with added supportive supplements, insulin, and antibiotics and, notably, without an extracellular matrix substitute or exogenous growth factors [
61,
62]. These organoids formed small, round spheres by 1 week and cell-dense organoids within 2–4 weeks [
61,
62]. The authors demonstrated that these GBM organoids recapitulated many characteristics intrinsic to human tumors [
62]. Their GBM organoids displayed characteristic histologic and immunohistochemical hallmarks of GBM, such as hypoxia gradients, nuclear atypia, and molecular heterogeneity [
62,
86]. The presence of hypoxia gradients is a hallmark of GBM, as hypoxia-induced signaling through notch and calcineurin pathways is known to contribute to proliferation of GSCs and correlates with patient survival [
87,
88,
89]. RNA and exome sequencing of these organoids demonstrated similarities in gene expression, allele frequency of somatic variants, and similar copy number ratios between the parent tumor and resultant organoids [
62]. Intratumor heterogeneity was also preserved in subregion organoid samples, where there was differential expression of gain-of-function epidermal growth factor receptor vIII (EGFRvIII) mutations within single organoids [
62,
84,
90]. Furthermore, these GBM organoids retained the spatial heterogeneity and clustering of neoplastic and immune cells (T cells and macrophages) of their parental tumors, a unique feature that previous models lacked [
44,
62,
68].
Importantly, these GBM organoids exhibited comparable tumor biology to their parental tumors, with similar ex vivo responses to both tumor-specific (tyrosine kinase inhibition) and non-specific (radiation and temozolomide) treatments as compared to their parent tumors [
62]. However, the MGMT methylation status did not consistently predict the response to radiation and temozolomide in GBM organoids described by Jacob et al., as is seen in the clinic [
62,
91]. Lastly, the GBM organoids engrafted into immunodeficient mice within 1–3 months and recapitulated the invasive morphology of human GBM tumors [
62]. Moreover, organoid-derived xenografts displayed ipsilateral and contralateral hemispheric invasion, parent-tumor-dependent satellite phenotypes, and neoangiogenesis by host endothelial cells [
62]. Recent work by Golebiewska and colleagues (2020) also demonstrated that xenografted glioma organoids maintain numerous genetic glioma subtypes [
63]. Together, these results highlight the similarities of histologic and phenotypic properties of GBM organoids to respective parent GBMs, a significant advance from prior glioma organoid models [
56,
61,
62,
63,
68].
Despite the rapid progress and interval advances in GBM organoid models, numerous limitations and questions remain. First, low success rates in IDH1-mutant (66.7%) and recurrent GBM (75%) indicate additional methodological optimization is needed [
61,
62]. Further, no data in published research currently exist on the ability to create PDOs from low-grade gliomas, which uniformly progress to higher-grade gliomas (WHO grade III and IV), or non-glioma brain tumors [
15,
16]. While the immune microenvironment seems to persist in current GBM organoid models, the extent to which it is represented is still in question as we continue to understand GBM–immune interactions [
62,
92]. The immune microenvironment has historically been difficult to demonstrate in vitro, even in organoid models where most immune cells have not been captured except when artificially replaced [
93]. It is also unclear whether nerve–tumor interactions are retained in organoid models, which are a known driver of high-grade glioma proliferation [
94]. Data are also underpowered in some experiments, limiting the ability to make definitive conclusions on the success of xenografts for different tumor subtypes and clinical correlation of therapeutic responses [
62,
63]. Further, glioblastoma is also marked by migration along blood vessels, which has not been demonstrated to date in current xenograft data [
95]. Despite the need to address these uncertainties, the existing evidence suggests that patient-derived glioma organoids may offer certain advantages over standard patient-derived cancer models.
This entry is adapted from the peer-reviewed paper 10.3390/jpm11050423