Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal solid tumors, associated with poor survival. The early stages usually lack symptoms, leading to a late diagnosis (stages III or IV) with limited therapeutic options and a dismal prognosis. Moreover, the metastatic potential of PDAC is extremely high and tumors spread mainly through lymphatic and blood vessels. Most of the patients already have metastasis in the liver and lymph nodes at the time of diagnosis. Epigenomic studies revealed that epigenetic changes in oncogenes and tumor suppressor genes were associated with epithelial-mesenchymal transition (EMT) which is responsible for the invasive phenotype of cancer cells and therefore, their metastatic potential.
- pancreatic ductal adenocarcinoma
- epigenetics
- epithelial–mesenchymal transition
The extensive research in genetics and genome-wide expression patterns indicates that genetic changes are critical for PDAC initiation and early progression. However, epigenetic alterations affected tumor progression and were associated with patients´ survival [1]. Epigenetic changes are heritable modifications of DNA or chromatin structures, which influence gene expression without altering the DNA sequence [2]. Epigenomics of PDAC studies the epigenetic changes across multiple genes or the entire genome. DNA methylation, post-translational modifications of histone proteins, and non-coding RNAs are the main epigenetic mechanisms that can influence gene expression. They have been confirmed as important contributors to PDAC development and progression [3][4][5].
EMT has emerged as a clinically relevant regulator of tumor cell spread to distant organs [6]. Early metastasis of PDAC results from dynamic gene expression changes, allowing epithelial tumor cells to acquire mesenchymal, fibroblast-like properties, increased migratory capacity, invasiveness, and resistance to therapy [7]. A cross-talk between individual counterparts of tumor microenvironment and innate and adaptive immune cells leads to PDAC progression (Figure 2).
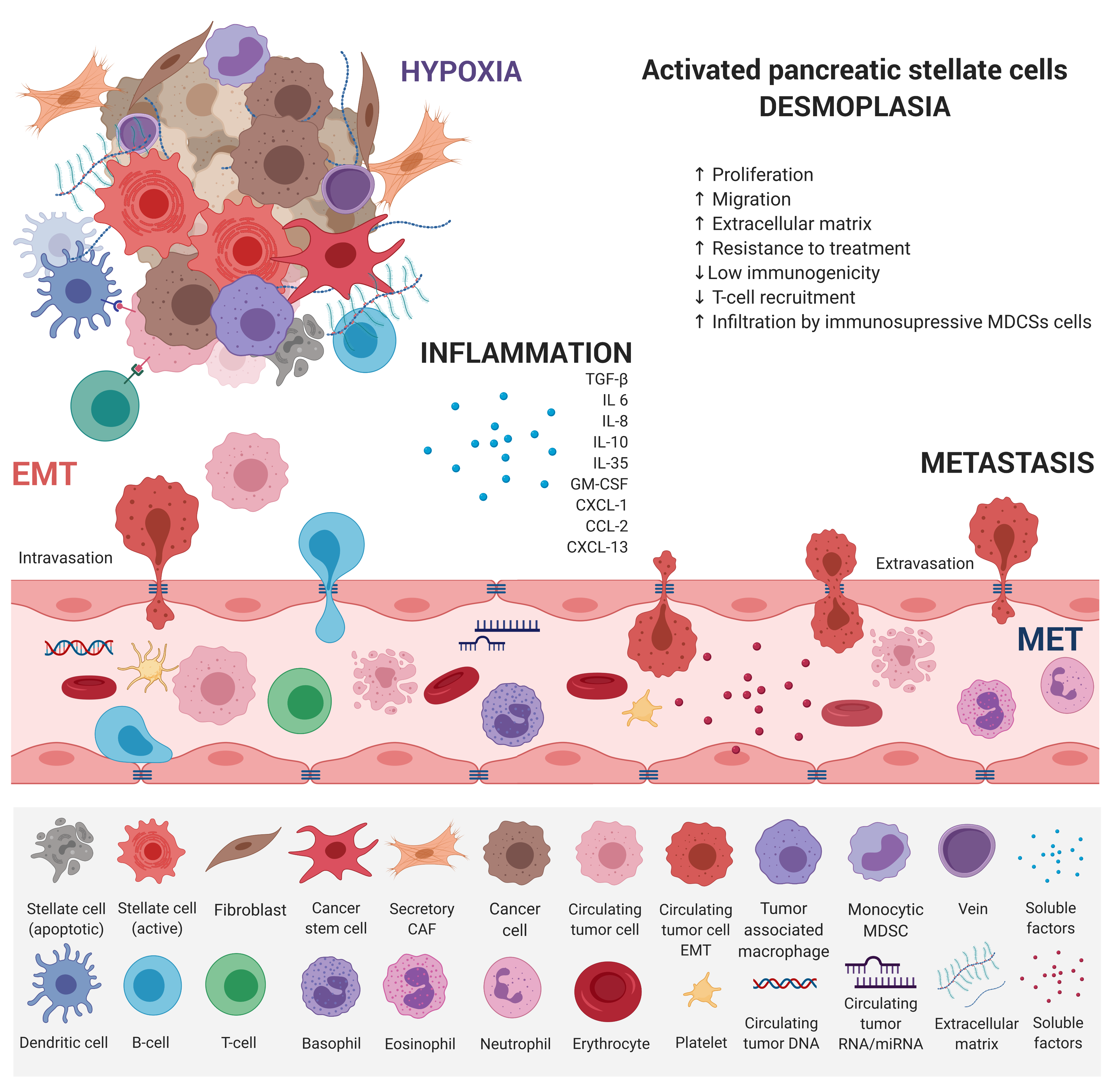
This dedifferentiation process is associated with a loss of functional epithelial cell markers, such as E-cadherin, and increased expression of mesenchymal markers [8]. Stem cell-associated marker expression of CD24+, CD44+, and CD133+ cells was found to correlate with EMT in PDAC [9]. TGF-b, TNF-a, as well as Notch, Hedgehog, and Wnt molecular pathways have been shown to induce EMT by the upregulation of Snail, Twist, and Zeb1 transcription factors, which destroy the tight junctions and degrades adhesion molecules [10][11][12][13][14][15][16]. Reversibility of EMT-related changes via epigenetic regulation allows the tumor cell to respond to various external and internal stimuli. Decreased expression of the CDH1 gene, which encodes the glycoprotein E-cadherin, is considered the hallmark of EMT, characterized by the dedifferentiation of epithelial cells and a loss of intercellular junctions[17]. Loss of E-cadherin-mediated cell-cell adhesion was associated with higher tumor invasiveness and increased metastatic potential [18]. This occurs in 42–60% of PDAC tumors and shows a strong correlation with metastasis to remote organs. However, a mechanism of CDH1 downregulation in most tumors is unknown. In fact, a decreased expression of the CDH1 gene is rarely induced by mutation or loss of heterozygosity. According to the published data, hypermethylation of the gene promoter region and interaction with the EMT-inducing transcription factors Slug, Snail, and Twist1 may explain the decreased CDH1 gene expression [19][20]. Moreover, von Burstin et al. demonstrated the downregulation of E-cadherin in metastatic PDAC cells mediated by a repressor complex, containing Snail and HDACs. The Snail protein binds directly to the CDH1 promoter, resulting in the inhibition of CDH1 expression, and Snail mRNA overexpression has been shown to correlate with the metastatic potential of cells [21]. These results were in accordance with previous findings showing an association between EMT and HDAC-mediated transcription control [22]. Several miRNAs were also identified as modulators of EMT, mainly the members of the miR-200 family and miR-205 [23], and their participation in PDAC tumors was also confirmed [24]. The role of epigenetic factors in EMT during malignant progression in PDAC was also confirmed by the epigenetic silencing of FOXA1/2 transcriptions factors, which were determined to be effective antagonists of EMT by promoter hypermethylation [25].
The success of PDAC treatment will be determined by the integration of genomic, epigenomic, and transcriptomic data into a biomarker-driven approach. Progress in early disease detection and reliable monitoring, together with a better understanding of EMT-associated epigenetic changes and the possibilities of their modulation will, hopefully soon, help to improve the outcome of patients with this devastating disease.
Acknowledgments: This research was funded by Scientific Grant Agency of the Ministry of Education, Science, Research and Sport of the Slovak Republic and Slovak Academy of Sciences VEGA, project number 2/0052/18 and by European Union´s Horizon 2020 grant agreement No 857381, project VISION (Strategies to strengthen scientific excellence and innovation capacity for early diagnosis of gastrointestinal cancers).
The article has been published on 10.3390/ijms21114091
References
- Thompson, M.J.; Rubbi, L.; Dawson, D.W.; Donahue, T.R.; Pellegrini, M.; Pancreatic Cancer Patient Survival Correlates with DNA Methylation of Pancreas Development Genes. PLOS ONE 2015, 10, e0128814, doi:10.1371/journal.pone.0128814.
- Liu, Z.; Gao, Y.; Li, X.; Cancer epigenetics and the potential of epigenetic drugs for treating solid tumors. Expert Rev Anticancer Ther 2019, 19, 139-149, doi:10.1080/14737140.2019.1552139.
- Henriksen, S.D.; Madsen, P.H.; Krarup, H.; Thorlacius-Ussing, O.; DNA Hypermethylation as a Blood-Based Marker for Pancreatic Cancer: A Literature Review. Pancreas 2015, 44, 1036-1045, doi:10.1097/mpa.0000000000000487.
- Syren, P.; Andersson, R.; Bauden, M.; Ansari, D.; Epigenetic alterations as biomarkers in pancreatic ductal adenocarcinoma. Scand J Gastroenterol 2017, 52, 668-673, doi:10.1080/00365521.2017.1301989.
- Tchio Mantho, C.I.; Harbuzariu, A.; Gonzalez-Perez, R.R.; Histone deacetylases, microRNA and leptin crosstalk in pancreatic cancer. World J Clin Oncol 2017, 8, 178-189, doi:10.5306/wjco.v8.i3.178.
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P., et al.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol 2017, 19, 518-529, doi:10.1038/ncb3513.
- Neureiter, D.; Jäger, T.; Ocker, M.; Kiesslich, T.; Epigenetics and pancreatic cancer: pathophysiology and novel treatment aspects. World journal of gastroenterology 2014, 20, 7830-7848, doi:10.3748/wjg.v20.i24.7830.
- Zagorac, S.; Garcia-Bermejo, L.; Sainz, B.; The Epigenetic Landscape of Pancreatic Cancer Stem Cells. Epigenomes 2018, 2, 10, .
- Zhang, Y.; Wei, J.; Wang, H.; Xue, X.; An, Y.; Tang, D.; Yuan, Z.; Wang, F.; Wu, J.; Zhang, J.; et al. Epithelial mesenchymal transition correlates with CD24+ CD44+ and CD133+ cells in pancreatic cancer. Oncology reports 2012, 27, 1599-1605, doi:10.3892/or.2012.1681.
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F.; Cancer-related inflammation. Nature 2008, 454, 436-444, doi:10.1038/nature07205.
- Klymkowsky, M.W.; Savagner, P.; Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am J Pathol 2009, 174, 1588-1593, doi:10.2353/ajpath.2009.080545.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A.; Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871-890, doi:10.1016/j.cell.2009.11.007.
- Voulgari, A.; Pintzas, A.; Epithelial–mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 2009, 1796, 75-90, doi:https://doi.org/10.1016/j.bbcan.2009.03.002.
- Vetter, G.; Saumet, A.; Moes, M.; Vallar, L.; Le Béchec, A.; Laurini, C.; Sabbah, M.; Arar, K.; Theillet, C.; Lecellier, C.H., et al.; et al. miR-661 expression in SNAI1-induced epithelial to mesenchymal transition contributes to breast cancer cell invasion by targeting Nectin-1 and StarD10 messengers. Oncogene 2010, 29, 4436-4448, doi:10.1038/onc.2010.181.
- Kotsafti, A.; Farinati, F.; Cardin, R.; Cillo, U.; Nitti, D.; Bortolami, M.; Autophagy and apoptosis-related genes in chronic liver disease and hepatocellular carcinoma. BMC gastroenterology 2012, 12, 118, doi:10.1186/1471-230X-12-118.
- Liu, J.; Hu, G.; Chen, D.; Gong, A.; Soori, G.; Dobleman, T.; Chen, X.-M.; Suppression of SCARA5 by Snail1 is essential for EMT-associated cell migration of A549 cells. Oncogenesis 2013, 2, e73-e73, doi:10.1038/oncsis.2013.37.
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J.; Direct regulation of TWIST by HIF-1α promotes metastasis. Nature cell biology 2008, 10, 295-305, doi:10.1038/ncb1691.
- Christofori, G.; Semb, H.; The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends in biochemical sciences 1999, 24, 73-76, doi:10.1016/s0968-0004(98)01343-7.
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A; Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer research 2008, 68, 3645-3654, doi:10.1158/0008-5472.CAN-07-2938.
- Peinado, H.; Olmeda, D.; Cano, A.; Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? . Nature reviews cancer 2007, 7, 415-428, doi:10.1038/nrc2131.
- von Burstin, J.; Eser, S.; Paul, M.C.; Seidler, B.; Brandl, M.; Messer, M.; von Werder, A.; Schmidt, A.; Mages, J.; Pagel, P., et al.; et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology 2009, 137, 361-71, 371.e1-5, doi:10.1053/j.gastro.2009.04.004.
- Glozak, M.A.; Seto, E.; Histone deacetylases and cancer. Oncogene 2007, 26, 5420-5432, doi:10.1038/sj.onc.1210610.
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J.; The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008, 10, 593-601, doi:10.1038/ncb1722.
- Paik, W.H.; Song, B.J.; Kim, H.W.; Kim, H.R.; Hwang, J.H.; MicroRNA-200c as a Prognostic Biomarker for Pancreatic Cancer. Korean J Gastroenterol 2015, 66, 215-220, doi:10.4166/kjg.2015.66.4.215.
- Song, Y.; Washington, M.K.; Crawford, H.C.; Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res 2010, 70, 2115-2125, doi:10.1158/0008-5472.can-09-2979.