Protein phosphorylation is a fundamental mechanism for many intracellular processes underlying cell life. This reversible mechanism, which is triggered by intra- and extra-cellular signals, regulates metabolism, transcription, proliferation, differentiation, cell movements, and apoptosis in countless cellular functions. Protein kinases form an enzyme family that catalyzes the transfer of the gamma-phosphate of adenosine triphosphate (ATP) to specific hydroxyl amino acids in protein substrates. On the other hand, protein phosphatases regulate the action of kinases, playing the role of regulators in the phosphorylation processes. The present investigation summarize the current knowledge on the roles playd by phosphatases in Chronic Myeloid Leukemia (CML).
- phosphatases
- kinases
- CML
- leukemia
- stem cell
1. Introduction
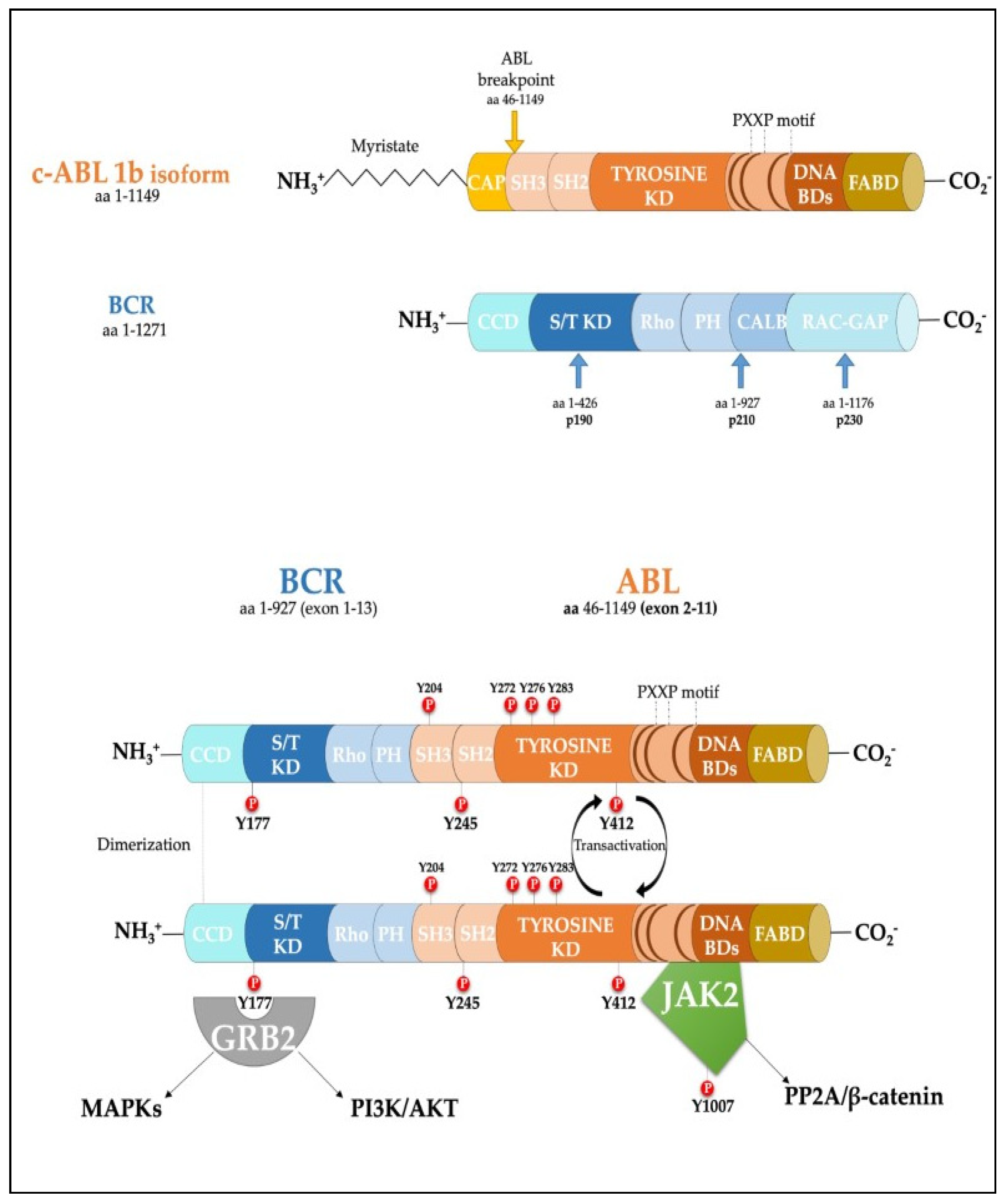 Figure 1. Scheme of the protein products derived from the BCR, ABL1b genes and the fusion product of the BCR-ABL1 oncogene. Major tyrosine phosphorylation sites are represented together with two well characterized interactors relevant for CML disease. SH Src homology domain; PTB Phosphotyrosine-binding domains; KD Kinase Domain; PH Pleckstrin homology domain; BD Binding Domain; FABD f-actin binding domain; CCD Coiled-coil domain.
Figure 1. Scheme of the protein products derived from the BCR, ABL1b genes and the fusion product of the BCR-ABL1 oncogene. Major tyrosine phosphorylation sites are represented together with two well characterized interactors relevant for CML disease. SH Src homology domain; PTB Phosphotyrosine-binding domains; KD Kinase Domain; PH Pleckstrin homology domain; BD Binding Domain; FABD f-actin binding domain; CCD Coiled-coil domain.2. Role of Phosphatases in the Regulation of Cell Proliferation
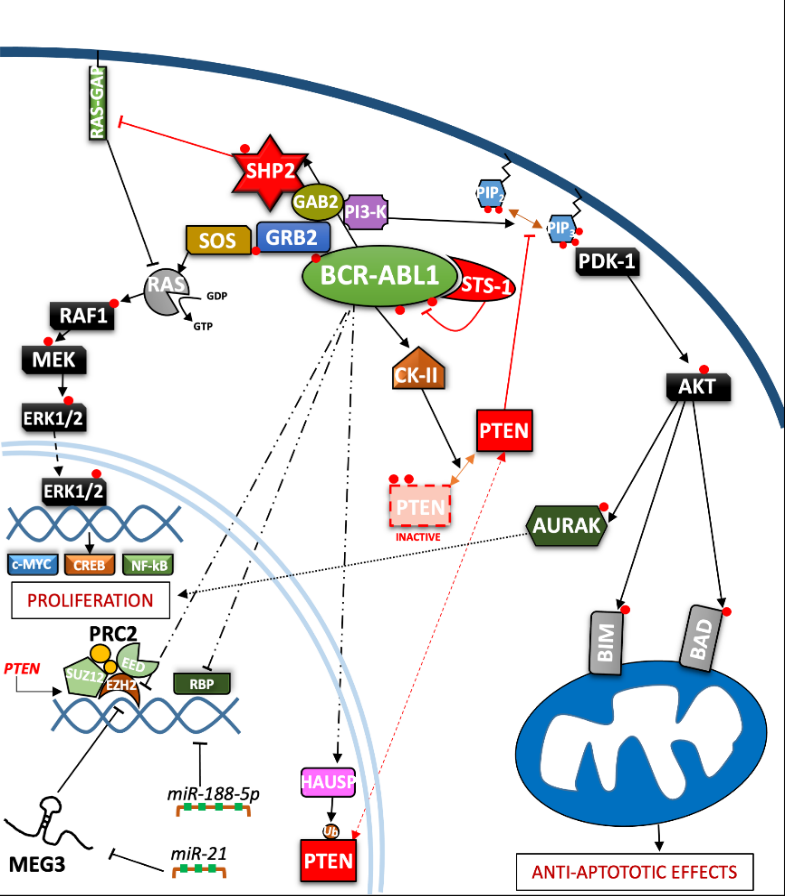
Figure 2. Depiction of the BCR-ABL1 kinase-driven proliferative processes in the context of CML. ERK = Extracellular signal-regulated kinases; MEG3 = Maternally expressed gene 3; PRC2 = Polycomb repressive complex 2; EZH = Enhancer of zeste polycomb repressive complex sub-unit.
| Protein | Coding Gene | Role in CML | References |
|---|---|---|---|
| Data verified in primary CML cells or in leukemia mouse models | |||
| DUSP1 | DUSP1 | Implicated in TKI-response | [67] |
| STS-1 | UBASH3B | Decreases cell proliferation Direct BCR-ABL1 regulation |
[25][23] |
| FAP-1 | PTPN13 | Regulation of β-catenin functions Decreases TKI sensitivity |
[68][69][70][71][72][73] |
| PTPRG | PTPRG | Regulation of β-catenin functions Implicated in TKI response |
[74][75][76][77][78][79][80][81][82] |
| SHP1 | PTPN6 | Acts through the PP2A on BCR-ABL1 Regulates BCR-ABL1-independent IM resistance |
[39][83][84][85][86][87] |
| SHP2 | PTPN11 | Increases cell proliferation Implicated in TKI resistance |
[30][32][33][34][35][36][37][38][39] |
| PP2A | PPP2CA | Quiescence and Self-renewal regulation Governs TKI-response |
[83][84][88][89][90][91][80][92][93][94][87][95][96][97][98] |
| PTEN | PTEN | Control of cell proliferation | [44][54][55][56][57][58][59][61][63][64][66] |
| Data obtained only in CML cell lines | |||
| PTP1B | PTPN1 | Reduces cell viability Correlated with IM response |
[99][100][101][102][103][104] |
| LAR –LIPRIN Α1 | PPFIA1 | Mitigates BCR-ABL1 leukemogenesis | [105] |
| LYP | PTPN22 | Decreases IM sensitivity | [106] |
| LMW-PTP | ACP1 | Regulates autophagy process Correlated with IM resistance |
[107][108] |
| PP1Α | PPP1CA | Improves cell survival and apoptosis resistance | [109] |
| TC45/TC48 | TC-PTP | Implicated in IM- and INFα-resistance Regulation of proliferation and apoptosis |
[110][111][112] |
This entry is adapted from the peer-reviewed paper 10.3390/cancers13102311
References
- Dan R Robinson; Yi-Mi Wu; Su-Fang Lin; The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548-5557, 10.1038/sj.onc.1203957.
- Mark A. Lemmon; Joseph Schlessinger; Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117-1134, 10.1016/j.cell.2010.06.011.
- Neel H. Shah; Jeanine F. Amacher; Laura M. Nocka; John Kuriyan; The Src module: an ancient scaffold in the evolution of cytoplasmic tyrosine kinases. Critical Reviews in Biochemistry and Molecular Biology 2018, 53, 535-563, 10.1080/10409238.2018.1495173.
- Sheila M. Thomas; Joan S. Brugge; CELLULAR FUNCTIONS REGULATED BY SRC FAMILY KINASES. Annual Review of Cell and Developmental Biology 1997, 13, 513-609, 10.1146/annurev.cellbio.13.1.513.
- Robert Roskoski; A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacological Research 2015, 100, 1-23, 10.1016/j.phrs.2015.07.010.
- Peter Blume-Jensen; Tony Hunter; Oncogenic kinase signalling. Nature 2001, 411, 355-365, 10.1038/35077225.
- Bhushan Nagar; Oliver Hantschel; Markus Seeliger; Jason M. Davies; William I. Weis; Giulio Superti-Furga; John Kuriyan; Organization of the SH3-SH2 Unit in Active and Inactive Forms of the c-Abl Tyrosine Kinase. Molecular Cell 2006, 21, 787-798, 10.1016/j.molcel.2006.01.035.
- Karel Dorey; John R Engen; Jana Kretzschmar; Matthias Wilm; Gitte Neubauer; Thomas Schindler; Giulio Superti-Furga; Phosphorylation and structure-based functional studies reveal a positive and a negative role for the activation loop of the c-Abl tyrosine kinase. Oncogene 2001, 20, 8075-8084, 10.1038/sj.onc.1205017.
- J R McWhirter; D L Galasso; J Y Wang; A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins.. Molecular and Cellular Biology 1993, 13, 7587-7595, 10.1128/mcb.13.12.7587.
- G Q Daley; R A Van Etten; D Baltimore; Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 1990, 247, 824-830, 10.1126/science.2406902.
- Brian J. Druker; Translation of the Philadelphia chromosome into therapy for CML. Blood 2008, 112, 4808-4817, 10.1182/blood-2008-07-077958.
- Jane F Apperley; Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. The Lancet Oncology 2007, 8, 1018-1029, 10.1016/s1470-2045(07)70342-x.
- Susan Branford; Zbigniew Rudzki; Sonya Walsh; Ian Parkinson; Andrew Grigg; Jeff Szer; Kerry Taylor; Richard Herrmann; John F. Seymour; Chris Arthur; et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood 2003, 102, 276-283, 10.1182/blood-2002-09-2896.
- Jorge Cortes; Elias Jabbour; Hagop Kantarjian; C. Cameron Yin; Jianqin Shan; Susan O'brien; Guillermo Garcia-Manero; Francis Giles; Megan Breeden; Nubia Reeves; et al. Dynamics of BCR-ABL kinase domain mutations in chronic myeloid leukemia after sequential treatment with multiple tyrosine kinase inhibitors. Blood 2007, 110, 4005-4011, 10.1182/blood-2007-03-080838.
- T P Hughes; G Saglio; A Quintás-Cardama; M J Mauro; D-W Kim; J H Lipton; M B Bradley-Garelik; J Ukropec; A Hochhaus; BCR-ABL1 mutation development during first-line treatment with dasatinib or imatinib for chronic myeloid leukemia in chronic phase. Leukemia 2015, 29, 1832-1838, 10.1038/leu.2015.168.
- David P. Labbé; Serge Hardy; Michel L. Tremblay; Protein Tyrosine Phosphatases in Cancer. Progress in Molecular Biology and Translational Science 2012, 2, 253-306, 10.1016/b978-0-12-396456-4.00009-2.
- Angela Bononi; Chiara Agnoletto; Elena De Marchi; Saverio Marchi; Simone Patergnani; Massimo Bonora; Carlotta Giorgi; Sonia Missiroli; Federica Poletti; Alessandro Rimessi; et al. Protein Kinases and Phosphatases in the Control of Cell Fate. Enzyme Research 2011, 2011, 1-26, 10.4061/2011/329098.
- Peter P. Ruvolo; Role of protein phosphatases in the cancer microenvironment. Biochimica et Biophysica Acta 2019, 1866, 144-152, 10.1016/j.bbamcr.2018.07.006.
- Mario Notari; Paolo Neviani; Ramasamy Santhanam; Bradley W. Blaser; Ji-Suk Chang; Annamaria Galietta; Anne E. Willis; Denis C. Roy; Michael A. Caligiuri; Guido Marcucci; et al. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood 2006, 107, 2507-2516, 10.1182/blood-2005-09-3732.
- Leonidas C. Platanias; Map kinase signaling pathways and hematologic malignancies. Blood 2003, 101, 4667-4679, 10.1182/blood-2002-12-3647.
- Chi-Dug Kang; Seok-Dong Yoo; Byung-Wook Hwang; Kwang-Woon Kim; Dong-Wan Kim; Cheol-Min Kim; Sun-Hee Kim; Byung-Seon Chung; The inhibition of ERK/MAPK not the activation of JNK/SAPK is primarily required to induce apoptosis in chronic myelogenous leukemic K562 cells. Leukemia Research 2000, 24, 527-534, 10.1016/s0145-2126(00)00010-2.
- Dejan Juric; Norman J. Lacayo; Meghan C. Ramsey; Janis Racevskis; Peter H. Wiernik; Jacob M. Rowe; Anthony H. Goldstone; Peter J. O'dwyer; Elisabeth Paietta; Branimir I. Sikic; et al. Differential Gene Expression Patterns and Interaction Networks in BCR-ABL–Positive and –Negative Adult Acute Lymphoblastic Leukemias. Journal of Clinical Oncology 2007, 25, 1341-1349, 10.1200/jco.2006.09.3534.
- J A Cutler; R Tahir; S K Sreenivasamurthy; C Mitchell; S Renuse; R S Nirujogi; A H Patil; M Heydarian; X Wong; X Wu; et al. Differential signaling through p190 and p210 BCR-ABL fusion proteins revealed by interactome and phosphoproteome analysis. Leukemia 2017, 31, 1513-1524, 10.1038/leu.2017.61.
- Marc Brehme; Oliver Hantschel; Jacques Colinge; Ines Kaupe; Melanie Planyavsky; Thomas Köcher; Karl Mechtler; Keiryn L. Bennett; Giulio Superti-Furga; Charting the molecular network of the drug target Bcr-Abl. Proceedings of the National Academy of Sciences 2009, 106, 7414-7419, 10.1073/pnas.0900653106.
- Afsar A. Mian; Ines Baumann; Marcus Liebermann; Florian Grebien; Giulio Superti-Furga; Martin Ruthardt; Oliver G. Ottmann; Oliver Hantschel; The phosphatase UBASH3B/Sts-1 is a negative regulator of Bcr-Abl kinase activity and leukemogenesis. Leukemia 2019, 33, 2319-2323, 10.1038/s41375-019-0468-y.
- Ann Marie Pendergast; Lawrence A. Quilliam; Larry D. Cripe; Craig H. Bassing; Zonghan Dai; Nanxin Li; Andreas Batzer; Kelly M. Rabun; Channing J. Der; Joseph Schlessinger; et al. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell 1993, 75, 175-185, 10.1016/s0092-8674(05)80094-7.
- Martin Sattler; M.Golam Mohi; Yuri B Pride; Laura R Quinnan; Nicole A Malouf; Klaus Podar; Franck Gesbert; Hiromi Iwasaki; Shaoguang Li; Richard A Van Etten; et al. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell 2002, 1, 479-492, 10.1016/s1535-6108(02)00074-0.
- H Modi; L Li; S Chu; J Rossi; J-K Yee; R Bhatia; Inhibition of Grb2 expression demonstrates an important role in BCR–ABL-mediated MAPK activation and transformation of primary human hematopoietic cells. Leukemia 2010, 25, 305-312, 10.1038/leu.2010.257.
- Min Chen; Ali G. Turhan; Hongxia Ding; Qingcong Lin; Kun Meng; Xiaoyan Jiang; Targeting BCR-ABL+ stem/progenitor cells and BCR-ABL-T315I mutant cells by effective inhibition of the BCR-ABL-Tyr177-GRB2 complex. Oncotarget 2017, 8, 43662-43677, 10.18632/oncotarget.18216.
- Haihua Gu; Joanne C. Pratt; Steven J. Burakoff; Benjamin G. Neel; Cloning of p97/Gab2, the Major SHP2-Binding Protein in Hematopoietic Cells, Reveals a Novel Pathway for Cytokine-Induced Gene Activation. Molecular Cell 1998, 2, 729-740, 10.1016/s1097-2765(00)80288-9.
- Y. Zhang; Ernesto Diaz-Flores; Geqiang Li; Zhengqi Wang; Zizhen Kang; Eleonora Haviernikova; Sara Rowe; Cheng-Kui Qu; William Tse; Kevin M. Shannon; et al. Abnormal hematopoiesis in Gab2 mutant mice. Blood 2007, 110, 116-124, 10.1182/blood-2006-11-060707.
- Cheng-Kui Qu; Suzanne Nguyen; Jianzhu Chen; Gen-Sheng Feng; Requirement of Shp-2 tyrosine phosphatase in lymphoid and hematopoietic cell development. Blood 2001, 97, 911-914, 10.1182/blood.v97.4.911.
- Marie Dance; Alexandra Montagner; Jean-Pierre Salles; Armelle Yart; Patrick Raynal; The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cellular Signalling 2008, 20, 453-459, 10.1016/j.cellsig.2007.10.002.
- Ruchi Pandey; Mallika Saxena; Reuben Kapur; Role of SHP2 in hematopoiesis and leukemogenesis. Current Opinion in Hematology 2017, 24, 307-313, 10.1097/moh.0000000000000345.
- Michaela Scherr; Anuhar Chaturvedi; Karin Battmer; Iris Dallmann; Beate Schultheis; Arnold Ganser; Matthias Eder; Enhanced sensitivity to inhibition of SHP2, STAT5, and Gab2 expression in chronic myeloid leukemia (CML). Blood 2006, 107, 3279-3287, 10.1182/blood-2005-08-3087.
- Rongzhen Xu; Yingzi Yu; Shu Zheng; Xiaoying Zhao; Qinghua Dong; Zhiwen He; Yun Liang; Qinghua Lu; Yongmin Fang; Xiaoxian Gan; et al. Overexpression of Shp2 tyrosine phosphatase is implicated in leukemogenesis in adult human leukemia. Blood 2005, 106, 3142-3149, 10.1182/blood-2004-10-4057.
- Shengqing Gu; Wayne W. Chan; Golam Mohi; Joel Rosenbaum; Azin Sayad; Zhibin Lu; Carl Virtanen; Shaoguang Li; Benjamin G. Neel; Richard A. Van Etten; et al. Distinct GAB2 signaling pathways are essential for myeloid and lymphoid transformation and leukemogenesis by BCR-ABL1. Blood 2016, 127, 1803-1813, 10.1182/blood-2015-06-653006.
- Shengqing Gu; Azin Sayad; Gordon Chan; Wentian Yang; Zhibin Lu; Carl Virtanen; Richard A. Van Etten; Benjamin G. Neel; SHP2 is required for BCR-ABL1-induced hematologic neoplasia. Leukemia 2017, 32, 203-213, 10.1038/leu.2017.250.
- Nicola Esposito; Irene Colavita; Concetta Quintarelli; Agostino Rodeo Sica; Anna Lucia Peluso; Luigia Luciano; Marco Picardi; Luigi Del Vecchio; Tonia Buonomo; Timothy P. Hughes; et al. SHP-1 expression accounts for resistance to imatinib treatment in Philadelphia chromosome–positive cells derived from patients with chronic myeloid leukemia. Blood 2011, 118, 3634-3644, 10.1182/blood-2011-03-341073.
- Alexandra Montagner; Armelle Yart; Marie Dance; Bertrand Perret; Jean-Pierre Salles; Patrick Raynal; A Novel Role for Gab1 and SHP2 in Epidermal Growth Factor-induced Ras Activation. Journal of Biological Chemistry 2005, 280, 5350-5360, 10.1074/jbc.m410012200.
- Michael G. Kharas; David A. Fruman; ABL Oncogenes and Phosphoinositide 3-Kinase: Mechanism of Activation and Downstream Effectors. Cancer Research 2005, 65, 2047-2053, 10.1158/0008-5472.can-04-3888.
- Sarah K. Tasian; David T. Teachey; Susan R. Rheingold; Targeting the PI3K/mTOR Pathway in Pediatric Hematologic Malignancies. Frontiers in Oncology 2014, 4, 108, 10.3389/fonc.2014.00108.
- Marisa Juntilla; Vineet D. Patil; Marco Calamito; Rohan P. Joshi; Morris J. Birnbaum; Gary A. Koretzky; AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 2010, 115, 4030-4038, 10.1182/blood-2009-09-241000.
- Zi-Yuan Nie; Lin Yang; Xiao-Jun Liu; Zhan Yang; Gao-Shan Yang; Jing Zhou; Yan Qin; Jing Yu; Ling-Ling Jiang; Jin-Kun Wen; et al. Morin Inhibits Proliferation and Induces Apoptosis by Modulating the miR-188-5p/PTEN/AKT Regulatory Pathway in CML Cells. Molecular Cancer Therapeutics 2019, 18, 2296-2307, 10.1158/1535-7163.mct-19-0051.
- Yu Chen; Tongtong Wang; Jing Du; Yanchun Li; Xin Wang; Yi Zhou; Xingxing Yu; Weimin Fan; Qiaojuan Zhu; Xiangmin Tong; et al. The Critical Role of PTEN/PI3K/AKT Signaling Pathway in Shikonin-Induced Apoptosis and Proliferation Inhibition of Chronic Myeloid Leukemia. Cellular Physiology and Biochemistry 2018, 47, 981-993, 10.1159/000490142.
- T Skorski; P Kanakaraj; M Nieborowska-Skorska; Mariusz Z. Ratajczak; Sc Wen; G Zon; Am Gewirtz; B Perussia; B Calabretta; Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood 1995, 86, 726-736, 10.1182/blood.v86.2.726.bloodjournal862726.
- Tomasz Skorski; Alfonso Bellacosa; Margaret Nieborowska‐Skorska; Miroslaw Majewski; Robert Martinez; John K. Choi; Rossana Trotta; Pawel Wlodarski; Danilo Perrotti; Tung O. Chan; et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway.. The EMBO Journal 1997, 16, 6151-6161, 10.1093/emboj/16.20.6151.
- Jing Yang; Takayuki Ikezoe; Chie Nishioka; Keiko Udaka; Akihito Yokoyama; Bcr‐Abl activates AURKA and AURKB in chronic myeloid leukemia cells via AKT signaling. International Journal of Cancer 2013, 134, 1183-1194, 10.1002/ijc.28434.
- H G Hamzah; A Pierce; W A Stewart; C Peter Downes; Alexander Gray; A Irvine; E Spooncer; A D Whetton; Chronic myeloid leukemia CD34+ cells have elevated levels of phosphatidylinositol 3,4,5 trisphosphate (PtdIns(3,4,5)P3) and lack a PtdIns(3,4,5)P3 response to cytokines and chemotactic factors; effects reversed by imatinib. Leukemia 2005, 19, 1851-1853, 10.1038/sj.leu.2403919.
- K Schuster; J Zheng; A A Arbini; C C Zhang; P P Scaglioni; Selective targeting of the mTORC1/2 protein kinase complexes leads to antileukemic effects in vitro and in vivo. Blood Cancer Journal 2011, 1, e34-e34, 10.1038/bcj.2011.30.
- Pengliang Xin; Chuntuan Li; Yan Zheng; Qunyi Peng; Huifang Xiao; Yuanling Huang; Xiongpeng Zhu; Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with imatinib mesylate against chronic myelogenous leukemia cell lines. Drug Design, Development and Therapy 2017, ume11, 1115-1126, 10.2147/dddt.s132092.
- Anthony J. Trimboli; Carmen Z. Cantemir-Stone; Fu Li; Julie A. Wallace; Anand Merchant; Nicholas Creasap; John C. Thompson; Enrico Caserta; Hui Wang; Jean-Leon Chong; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084-1091, 10.1038/nature08486.
- C Nishioka; T Ikezoe; J Yang; A Yokoyama; Long-term exposure of leukemia cells to multi-targeted tyrosine kinase inhibitor induces activations of AKT, ERK and STAT5 signaling via epigenetic silencing of the PTEN gene. Leukemia 2010, 24, 1631-1640, 10.1038/leu.2010.145.
- Cong Peng; Yaoyu Chen; Zhongfa Yang; Haojian Zhang; Lori Osterby; Alan G. Rosmarin; Shaoguang Li; PTEN is a tumor suppressor in CML stem cells and BCR-ABL–induced leukemias in mice. Blood 2010, 115, 626-635, 10.1182/blood-2009-06-228130.
- Cristina Panuzzo; Sabrina Crivellaro; Giovanna Carra; Angelo Guerrasio; Giuseppe Saglio; Alessandro Morotti; BCR-ABL Promotes PTEN Downregulation in Chronic Myeloid Leukemia. PLOS ONE 2014, 9, e110682, 10.1371/journal.pone.0110682.
- C. Bassi; J. Ho; T. Srikumar; R. J. O. Dowling; C. Gorrini; S. J. Miller; T. W. Mak; B. G. Neel; B. Raught; Vuk Stambolic; et al. Nuclear PTEN Controls DNA Repair and Sensitivity to Genotoxic Stress. Science 2013, 341, 395-399, 10.1126/science.1236188.
- Min Sup Song; Leonardo Salmena; Arkaitz Carracedo; Ainara Egia; Francesco Lo-Coco; Julie Teruya-Feldstein; Pier Paolo Pandolfi; The deubiquitinylation and localization of PTEN are regulated by a HAUSP–PML network. Nature 2008, 455, 813-817, 10.1038/nature07290.
- A Morotti; C Panuzzo; S Crivellaro; B Pergolizzi; U Familiari; A H Berger; G Saglio; Pier Paolo Pandolfi; BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia 2013, 28, 1326-1333, 10.1038/leu.2013.370.
- Alessandro Morotti; Cristina Panuzzo; Sabrina Crivellaro; Giovanna Carrà; Carmen Fava; Angelo Guerrasio; Pier Paolo Pandolfi; Giuseppe Saglio; BCR-ABL inactivates cytosolic PTEN through Casein Kinase II mediated tail phosphorylation. Cell Cycle 2015, 14, 973-979, 10.1080/15384101.2015.1006970.
- Raphaël Margueron; Danny Reinberg; The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343-349, 10.1038/nature09784.
- Huafeng Xie; Cong Peng; Jialiang Huang; Bin E. Li; Woojin Kim; Elenoe C. Smith; Yuko Fujiwara; Jun Qi; Giulia Cheloni; Partha P. Das; et al. Chronic Myelogenous Leukemia– Initiating Cells Require Polycomb Group Protein EZH2. Cancer Discovery 2016, 6, 1237-1247, 10.1158/2159-8290.cd-15-1439.
- Mary T. Scott; Koorosh Korfi; Peter Saffrey; Lisa E.M. Hopcroft; Ross Kinstrie; Francesca Pellicano; Carla Guenther; Paolo Gallipoli; Michelle Cruz; Karen Dunn; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discovery 2016, 6, 1248-1257, 10.1158/2159-8290.cd-16-0263.
- Jingfeng Zhou; Danian Nie; Juan Li; Xin Du; Yuhong Lu; Yangqiu Li; Chang Liu; Wei Dai; Yun Wang; Yanli Jin; et al. PTEN Is Fundamental for Elimination of Leukemia Stem Cells Mediated by GSK126 Targeting EZH2 in Chronic Myelogenous Leukemia. Clinical Cancer Research 2017, 24, 145-157, 10.1158/1078-0432.ccr-17-1533.
- Xiangyu Zhou; Ping Yuan; Qi Liu; Zhiqiang Liu; LncRNA MEG3 Regulates Imatinib Resistance in Chronic Myeloid Leukemia via Suppressing MicroRNA-21. Biomolecules & Therapeutics 2017, 25, 490-496, 10.4062/biomolther.2016.162.
- Ziye Li; Lin Yang; Xiaojun Liu; Ziyuan Nie; Jianmin Luo; Long noncoding RNA MEG3 inhibits proliferation of chronic myeloid leukemia cells by sponging microRNA21. Biomedicine & Pharmacotherapy 2018, 104, 181-192, 10.1016/j.biopha.2018.05.047.
- Xiaolin Yin; Minran Zhou; Yue Fu; Lin Yang; Man Xu; Ting Sun; XiaoMing Wang; Tao Huang; Chunyan Chen; Histone demethylase RBP2 mediates the blast crisis of chronic myeloid leukemia through an RBP2/PTEN/BCR-ABL cascade.. Cellular Signalling 2019, 63, 109360, 10.1016/j.cellsig.2019.109360.
- Kesarwani, M.; Kincaid, Z.; Gomaa, A.; Huber, E.; Rohrabaugh, S.; Siddiqui, Z.; Bouso, M.F.; Latif, T.; Xu, M.; Komurov, K.; et al. Targeting c-FOS and DUSP1 abrogates intrinsic resistance to tyrosine-kinase inhibitor therapy in BCR-ABL-induced leukemia. Nat. Med. 2017, 23, 472–482.
- Huang, W.; Zhu, C.; Wang, H.; Horvath, E.; Eklund, E.A. The Interferon Consensus Sequence-binding Protein (ICSBP/IRF8) Represses PTPN13 Gene Transcription in Differentiating Myeloid Cells. J. Biol. Chem. 2008, 283, 7921–7935.
- Hao, S.X.; Ren, R.; Jahn, T.; Seipel, P.; Urschel, S.; Peschel, C.; Duyster, J. Expression of Interferon Consensus Sequence Binding Protein (ICSBP) Is Downregulated in Bcr-Abl-Induced Murine Chronic Myelogenous Leukemia-Like Disease, and Forced Coexpression of ICSBP Inhibits Bcr-Abl-Induced Myeloproliferative Disorder. Mol. Cell. Biol. 2000, 20, 979–991.
- Schmidt, M.; Nagel, S.; Proba, J.; Thiede, C.; Ritter, M.; Waring, J.F.; Rosenbauer, F.; Huhn, D.; Wittig, B.; Horak, I.; et al. Lack of interferon consensus sequence binding protein (ICSBP) transcripts in human myeloid leukemias. Blood 1998, 91, 22–29.
- Huang, W.; Bei, L.; Eklund, E.A. Fas-associated phosphatase 1 (Fap1) influences betacatenin activity in myeloid progenitor cells expressing the Bcr-abl oncogene. J. Biol. Chem. 2013, 288, 12766–12776.
- Huang, W.; Bei, L.; Eklund, E.A. Fas-associated phosphatase 1 mediates Fas resistance in myeloid progenitor cells expressing the Bcr–abl oncogene. Leuk. Lymphoma 2012, 54, 619–630.
- Huang, W.; Luan, C.-H.; Hjort, E.E.; Bei, L.; Mishra, R.; Sakamoto, K.M.; Platanias, L.C.; Eklund, E.A. The role of Fas-associated phosphatase 1 in leukemia stem cell persistence during tyrosine kinase inhibitor treatment of chronic myeloid leukemia. Leukemia 2016, 30, 1502–1509.
- Lissandrini, D.; Vermi, W.; Vezzalini, M.; Sozzani, S.; Facchetti, F.; Bellone, G.; Mafficini, A.; Gentili, F.; Ennas, M.G.; Tecchio, C.; et al. Receptor-type protein tyrosine phosphatase gamma (PTPγ), a new identifier for myeloid dendritic cells and specialized macrophages. Blood 2006, 108, 4223–4231.
- Sorio, C.; Melotti, P.; D’Arcangelo, D.; Mendrola, J.; Calabretta, B.; Croce, C.M.; Huebner, K. Receptor protein tyrosine phosphatase gamma, Ptp gamma, regulates hematopoietic differentiation. Blood 1997, 90, 49–57.
- Mafficini, A.; Vezzalini, M.; Zamai, L.; Galeotti, L.; Bergamini, G.; Della Peruta, M.; Melotti, P.; Sorio, C. Protein Tyrosine Phosphatase Gamma (PTPgamma) is a Novel Leukocyte Marker Highly Expressed by CD34 Precursors. Biomark. Insights 2007, 2, 218–225.
- Della Peruta, M.; Martinelli, G.; Moratti, E.; Pintani, D.; Vezzalini, M.; Mafficini, A.; Grafone, T.; Iacobucci, I.; Soverini, S.; Murineddu, M.; et al. Protein tyrosine phosphatase receptor type is a functional tumor suppressor gene specifically downregulated in chronic myeloid leukemia. Cancer Res. 2010, 70, 8896–8906.
- Vezzalini, M.; Mafficini, A.; Tomasello, L.; Lorenzetto, E.; Moratti, E.; Fiorini, Z.; Holyoake, T.L.; Pellicano, F.; Krampera, M.; Tecchio, C.; et al. A new monoclonal antibody detects downregulation of protein tyrosine phosphatase receptor type gamma in chronic myeloid leukemia patients. J. Hematol. Oncol. 2017, 10, 129.
- Tomasello, L.; Vezzalini, M.; Boni, C.; Bonifacio, M.; Scaffidi, L.; Yassin, M.; Al-Dewik, N.; Takam Kamga, P.; Krampera, M.; Sorio, C. Regulative Loop between beta-catenin and Protein Tyrosine Receptor Type gamma in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 2298.
- Ismail, M.A.; Samara, M.; Al Sayab, A.; Alsharshani, M.; Yassin, M.A.; Varadharaj, G.; Vezzalini, M.; Tomasello, L.; Monne, M.; Morsi, H.; et al. Aberrant DNA methylation of PTPRG as one possible mechanism of its under-expression in CML patients in the State of Qatar. Mol. Genet. Genom. Med. 2020, 8, 1319.
- Ismail, M.A.; Vezzalini, M.; Morsi, H.; Abujaber, A.; Al Sayab, A.; Siveen, K.; Yassin, M.A.; Monne, M.; Samara, M.; Cook, R.; et al. Predictive value of tyrosine phosphatase receptor gamma for the response to treatment tyrosine kinase inhibitors in chronic myeloid leukemia patients. Sci. Rep. 2021, 11, 8833.
- Drube, J.; Ernst, T.; Pfirrmann, M.; Albert, B.V.; Drube, S.; Reich, D.; Kresinsky, A.; Halfter, K.; Sorio, C.; Fabisch, C.; et al. PTPRG and PTPRC modulate nilotinib response in chronic myeloid leukemia cells. Oncotarget 2018, 9, 9442–9455.
- Neviani, P.; Santhanam, R.; Trotta, R.; Notari, M.; Blaser, B.W.; Liu, S.; Mao, H.; Chang, J.S.; Galietta, A.; Uttam, A.; et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell 2005, 8, 355–368.
- Samanta, A.K.; Chakraborty, S.N.; Wang, Y.; Kantarjian, H.; Sun, X.; Hood, J.; Perrotti, D.; Arlinghaus, R.B. Jak2 inhibition deactivates Lyn kinase through the SET–PP2A–SHP1 pathway, causing apoptosis in drug-resistant cells from chronic myelogenous leukemia patients. Oncogene 2009, 28, 1669–1681.
- Li, Y.; Liu, X.; Guo, X.; Liu, X.; Luo, J. DNA methyltransferase 1 mediated aberrant methylation and silencing of SHP-1 gene in chronic myelogenous leukemia cells. Leuk. Res. 2017, 58, 9–13.
- Zhang, X.; Yang, L.; Liu, X.; Nie, Z.; Wang, X.; Pan, Y.; Luo, J. Research on the epigenetic regulation mechanism of thePTPN6gene in advanced chronic myeloid leukaemia. Br. J. Haematol. 2017, 178, 728–738.
- Salas, A.; Ponnusamy, S.; Senkal, C.E.; Meyers-Needham, M.; Selvam, S.P.; Saddoughi, S.A.; Apohan, E.; Sentelle, R.D.; Smith, C.; Gault, C.R.; et al. Sphingosine kinase-1 and sphingosine 1-phosphate receptor 2 mediate Bcr-Abl1 stability and drug resistance by modulation of protein phosphatase 2A. Blood 2011, 117, 5941–5952.
- Neviani, P.; Santhanam, R.; Oaks, J.J.; Eiring, A.M.; Notari, M.; Blaser, B.W.; Liu, S.; Trotta, R.; Muthusamy, N.; Gambacorti-Passerini, C.; et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome–positive acute lymphocytic leukemia. J. Clin. Investig. 2007, 117, 2408–2421.
- Lucas, C.M.; Harris, R.J.; Giannoudis, A.; Copland, M.; Slupsky, J.R.; Clark, R.E. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood 2011, 117, 6660–6668.
- Lucas, C.M.; Milani, M.; Butterworth, M.; Carmell, N.; Scott, L.J.; Clark, R.E.; Cohen, G.M.; Varadarajan, S. High CIP2A levels correlate with an antiapoptotic phenotype that can be overcome by targeting BCL-XL in chronic myeloid leukemia. Leukemia 2016, 30, 1273–1281.
- Silvestri, G.; Trotta, R.; Stramucci, L.; Ellis, J.J.; Harb, J.G.; Neviani, P.; Wang, S.; Eisfeld, A.K.; Walker, C.J.; Zhang, B.; et al. Persistence of Drug-Resistant Leukemic Stem Cells and Impaired NK Cell Immunity in CML Patients Depend on MIR300 Antiproliferative and PP2A-Activating Functions. Blood Cancer Discov. 2020, 1, 48–67.
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 2013, 123, 4144–4157.
- Wang, S.; Xie, W.; Wang, D.; Peng, Z.; Zheng, Y.; Liu, N.; Dai, W.; Wang, Y.; Wang, Z.; Yang, Y.; et al. Discovery of a small molecule targeting SET-PP2A interaction to overcome BCR-ABLT315I mutation of chronic myeloid leukemia. Oncotarget 2015, 6, 12128–12140.
- Agarwal, A.; MacKenzie, R.J.; Pippa, R.; Eide, C.A.; Oddo, J.; Tyner, J.W.; Sears, R.; Vitek, M.P.; Odero, M.D.; Christensen, D.J.; et al. Antagonism of SET Using OP449 Enhances the Efficacy of Tyrosine Kinase Inhibitors and Overcomes Drug Resistance in Myeloid Leukemia. Clin. Cancer Res. 2014, 20, 2092–2103.
- Laidlaw, K.M.E.; Berhan, S.; Liu, S.; Silvestri, G.; Holyoake, T.L.; Frank, D.A.; Aggarwal, B.; Bonner, M.Y.; Perrotti, D.; Jørgensen, H.G.; et al. Cooperation of imipramine blue and tyrosine kinase blockade demonstrates activity against chronic myeloid leukemia. Oncotarget 2016, 7, 51651–51664.
- Lai, D.; Chen, M.; Su, J.; Liu, X.; Rothe, K.; Hu, K.; Forrest, D.L.; Eaves, C.J.; Morin, G.B.; Jiang, X. PP2A inhibition sensitizes cancer stem cells to ABL tyrosine kinase inhibitors in BCR-ABL+human leukemia. Sci. Transl. Med. 2018, 10, eaan8735.
- Lai, D.; Chen, M.; Su, J.; Liu, X.; Rothe, K.; Hu, K.; Forrest, D.L.; Eaves, C.J.; Morin, G.B.; Jiang, X. Response to Comment on “PP2A inhibition sensitizes cancer stem cells to ABL tyrosine kinase inhibitors in BCR-ABL+ human leukemia”. Sci. Transl. Med. 2019, 11, eaav0819.
- Perrotti, D.; Agarwal, A.; Lucas, C.M.; Narla, G.; Neviani, P.; Odero, M.D.; Ruvolo, P.P.; Verrills, N.M. Comment on “PP2A inhibition sensitizes cancer stem cells to ABL tyrosine kinase inhibitors in BCR-ABL human leukemia”. Sci. Transl. Med. 2019, 11, eaau0416.
- Través, P.G.; Pardo, V.; Pimentel-Santillana, M.; González-Rodríguez, Á.; Mojena, M.; Rico, D.; Montenegro, Y.; Calés, C.; Martín-Sanz, P.; Valverde, A.M.; et al. Pivotal role of protein tyrosine phosphatase 1B (PTP1B) in the macrophage response to pro-inflammatory and anti-inflammatory challenge. Cell Death Dis. 2014, 5, e1125.
- Le Sommer, S.; Morrice, N.; Pesaresi, M.; Thompson, D.; Vickers, M.A.; Murray, G.I.; Mody, N.; Neel, B.G.; Bence, K.K.; Wilson, H.M.; et al. Deficiency in Protein Tyrosine Phosphatase PTP1B Shortens Lifespan and Leads to Development of Acute Leukemia. Cancer Res. 2018, 78, 75–87.
- Alvira, D.; Naughton, R.; Bhatt, L.; Tedesco, S.; Landry, W.D.; Cotter, T.G. Inhibition of Protein-tyrosine Phosphatase 1B (PTP1B) Mediates Ubiquitination and Degradation of Bcr-Abl Protein. J. Biol. Chem. 2011, 286, 32313–32323.
- LaMontagne, K.R.; Hannon, G.; Tonks, N.K. Protein tyrosine phosphatase PTP1B suppresses p210 bcr-abl-induced transformation of Rat-1 fibroblasts and promotes differentiation of K562 cells. Proc. Natl. Acad. Sci. USA 1998, 95, 14094–14099.
- Elgehama, A.; Chen, W.; Pang, J.; Mi, S.; Li, J.; Guo, W.; Wang, X.; Gao, J.; Yu, B.; Shen, Y.; et al. Blockade of the interaction between Bcr-Abl and PTB1B by small molecule SBF-1 to overcome imatinib-resistance of chronic myeloid leukemia cells. Cancer Lett. 2016, 372, 82–88.
- Koyama, N.; Koschmieder, S.; Tyagi, S.; Portero-Robles, I.; Chromic, J.; Myloch, S.; Nürnberger, H.; Rossmanith, T.; Hofmann, W.-K.; Hoelzer, D.; et al. Inhibition of Phosphotyrosine Phosphatase 1B Causes Resistance in BCR-ABL-Positive Leukemia Cells to the ABL Kinase Inhibitor STI571. Clin. Cancer Res. 2006, 12, 2025–2031.
- Gu, C.; Liu, Y.; Yin, Z.; Yang, J.; Huang, G.; Zhu, X.; Li, Y.; Fei, J. Discovery of the Oncogenic Parp1, a Target of bcr-abl and a Potential Therapeutic, in mir-181a/PPFIA1 Signaling Pathway. Mol. Ther. Nucleic Acids 2019, 16, 1–14.
- Chien, W.; Tidow, N.; Williamson, E.A.; Shih, L.-Y.; Krug, U.; Kettenbach, A.; Fermin, A.C.; Roifman, C.M.; Koeffler, H. Characterization of a Myeloid Tyrosine Phosphatase, Lyp, and Its Role in the Bcr-Abl Signal Transduction Pathway. J. Biol. Chem. 2003, 278, 27413–27420.
- Faria, A.V.S.; Clerici, S.P.; de Souza Oliveira, P.F.; Queiroz, K.C.S.; Peppelenbosch, M.P.; Ferreira-Halder, C.V. LMWPTP modulates the antioxidant response and autophagy process in human chronic myeloid leukemia cells. Mol. Cell. Biochem. 2020, 466, 83–89.
- Ferreira, P.A.; Ruela-De-Sousa, R.R.; Queiroz, K.C.S.; Souza, A.C.S.; Milani, R.; Pilli, R.A.; Peppelenbosch, M.P.; Hertog, J.D.; Ferreira, C.V. Knocking Down Low Molecular Weight Protein Tyrosine Phosphatase (LMW-PTP) Reverts Chemoresistance through Inactivation of Src and Bcr-Abl Proteins. PLoS ONE 2012, 7, e44312.
- Naughton, R.; Quiney, C.; Turner, S.D.; Cotter, T.G. Bcr-Abl-mediated redox regulation of the PI3K/AKT pathway. Leukemia 2009, 23, 1432–1440.
- Shimizu, T.; Miyakawa, Y.; Oda, A.; Kizaki, M.; Ikeda, Y. STI571-resistant KT-1 cells are sensitive to interferon-α accompanied by the loss of T-cell protein tyrosine phosphatase and prolonged phosphorylation of Stat1. Exp. Hematol. 2003, 31, 601–608.
- Shimizu, T.; Miyakawa, Y.; Iwata, S.; Kuribara, A.; Tiganis, T.; Morimoto, C.; Ikeda, Y.; Kizaki, M. A novel mechanism for imatinib mesylate (STI571) resistance in CML cell line KT-1: Role of TC-PTP in modulating signals downstream from the BCR-ABL fusion protein. Exp. Hematol. 2004, 32, 1057–1063.
- Mitra, A.; Sasikumar, K.; Parthasaradhi, B.; Radha, V. The tyrosine phosphatase TC48 interacts with and inactivates the oncogenic fusion protein BCR-Abl but not cellular Abl. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 275–284.