Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cardiac & Cardiovascular Systems
The renin–angiotensin system (RAS) indicates its central role in the pathogenesis of cardiovascular remodelling via both hemodynamic alterations and direct growth and the proliferation effects of angiotensin II or aldosterone resulting in the hypertrophy of cardiomyocytes, the proliferation of fibroblasts, and inflammatory immune cell activation.
- miRNA
- RAS
- cardiac remodelling
- cardiac fibrosis
- cardiac hypertrophy
1. The Renin–Angiotensin System
The renin–angiotensin system (RAS) has been traditionally associated with the control of electrolyte balance and blood pressure regulation. However, its chronic activation may impair cardiac function and structure both via hemodynamic overload as well as direct trophic action on cardiac and vascular tissues. The deleterious effect of chronic RAS activation is reflected by the success of RAS inhibition in a variety of cardiovascular pathologies [1][2][3][4][5][6][7].
1.1. Classical Pathways
The classical pathway of the RAS is activated by the renin-induced cleavage of liver-synthesized angiotensinogen to the primary effector molecule, angiotensin II (Ang II). Ang II is the principal player in the RAS, whose effect is mainly mediated by binding to angiotensin receptor type I (AT1R); however, angiotensin receptor type II (AT2R) and other G protein-coupled receptors (GPCRs) may be involved [8]. The crucial Ang II effects are mediated by AT1R are circulatory volume and vascular tone regulation, hypertrophic and fibrotic target organ remodelling, reactive oxygen species (ROS) production, and the modulation of immune response and inflammation [9][10][11][12]. The function of AT2R is not fully understood, but it is considered to antagonize the effect of AT1R by promoting vasodilation along with antiproliferative, anti-fibrotic, and anti-inflammatory effects [8][13].
1.2. Alternative Pathways of RAS
ACE inhibition does not entirely inhibit Ang II biosynthesis, as other proteases such as kallikrein, cathepsin G, heart chymase, and elastase-2 also converting Ang I to Ang II [14][15].
Moreover, Ang I and Ang II are alternatively cleaved into Ang (1-9) and Ang (1-7), respectively, by the action of the ACE2 enzyme, although Ang II is significantly preferred over Ang I as a substrate [16][17][18]. Ang (1-7) can also be formed from Ang (1-9) by ACE or directly from Ang I by endopeptidases such as neprilysin, or from Ang II by prolylcarboxypeptidase [19]. Increased ACE2 expression attenuates Ang II-induced cardiac hypertrophy while its reduced expression is associated with impaired cardiac contractility and hypertension development [20][21]. The biological effects of Ang (1-7) counteract many of the effects of Ang II, as it promotes vasodilation while inhibiting proliferation, hypertrophy, fibrosis, thrombosis, and rhythm disorders [19][22][23].
The beneficial effects of ACE inhibitors and AT1R blockers (ARB) are at least partially mediated through the increased accumulation of Ang (1-7). ACE inhibition leads to Ang I accumulation and its increased conversion to Ang (1-7) [24]. On the other hand, the blockade of AT1R by ARB leads to the accumulation of Ang II, which is subsequently converted to Ang (1-7) by ACE2 [24]. The target receptor of Ang (1-7) is the Mas receptor (MasR) while the aspartate decarboxylated product of Ang II alamandine binds to the MAS1-related G protein-coupled receptor D (MrgD), both taking part in the protective arm of RAS along with AT2R [25][26][27][28]. Additionally, the octapeptide angioprotectin is a more effective MasR agonist than Ang (1-7), which exerts vasodilatory action [29][30].
Angiotensin I can also be cleaved by aminopeptidase A and ACE into angiotensin III (Ang III) (Ang 2-8) and subsequent cleavage by aminopeptidase N leads to the formation of angiotensin IV (Ang IV) (Ang 3-8). Ang III can bind to both AT1R and AT2R, while Ang IV acts on AT4R, also known as insulin-regulated aminopeptidases (IRAP), located predominantly in the brain but also in the heart [8][31]. Ang IV is primarily associated with improved cognitive and memory functions, as it dose-dependently inhibits IRAP and induces the accumulation of neuropeptides [8][32].
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) and the adipokine apelin, other alternative substrates of ACE and ACE2 respectively, also join the cardioprotective arm of the RAS. Ac-SDKP administration exerts an anti-inflammatory, antiproliferation, antidifferentiation and anti-migration effect which in general attenuates cardiac fibrosis and Ac-SDKP augmentation by ACE inhibitors and partially mediates their pharmacological benefits [33]. Apelin binds to the GPCR APJ, exerting positive inotropic as well as cardioprotective effects [34][35][36].
To sum up, the RAS involves various molecules with opposing biological effects. One line is represented by vasoconstrictive, pro-proliferative and pro-inflammatory molecules, such as ACE, Ang II, and AT1R. The other line involves ACE2, Ang (1-7), AT2R, MasR, MrgD, and APJ, which exert effects at least partially counteracting the potentially harmful actions of the deleterious arm of the RAS (Figure 1).
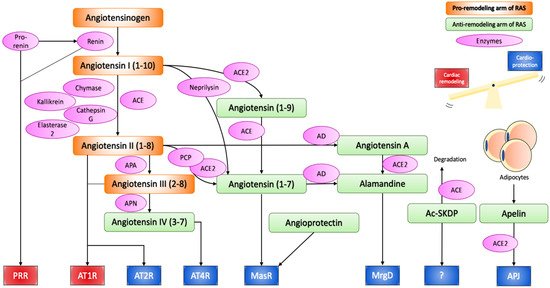
Figure 1. Renin–angiotensin system.
Angiotensinogen produced from the liver is proteolytically activated to angiotensin I and II through the action of renin, ACE, chymase, kallikrein, cathepsin G, and esterase-2. Angiotensin II can bind to AT1R and AT2R to initiate the effect of the RAS, or can be further cleaved or modified into cardioprotective peptides, namely angiotensin IV, angiotensin (1-7), and alamandine, which exert their effect through AT4R, MasR and MrgD respectively. Angioprotectin, Ac-SKDP, Apelin have also emerged as new cardioprotective peptides of the RAS. Thus, the RAS can induce both pathological cardiac remodelling and cardioprotective effects, and the balance between the two determines the overall effect on the cardiovascular system. In chronic activation of the RAS, the cardiac remodelling arm is often stronger, as suggested by the therapeutic success of ACE inhibitors and AT1R blockers.
ACE, angiotensin converting enzyme; ACE2, angiotensin converting enzyme 2; APA, aminopeptidase A; APN, aminopeptidase N; PCP, prolylcarboxypeptidase; AD, aspartate decarboxylase; PRR, (pro)renin receptor; AT1R, angiotensin II receptor type 1; AT2R, angiotensin II receptor type 2; MasR, mas receptor; MrgD, MAS1-related G protein-coupled receptor D; Ac-SKDP, N-acetyl-seryl-aspartyl-lysyl-proline.
1.3. Local RAS
RAS components can be synthesized at a local tissue scale by the peptidases of the alternative pathways, a process known as the local RAS, which has been recognized to play an important role in tissue remodelling [37][38]. The local RAS is also activated by the binding of renin or prorenin to the (pro)renin receptor (PRR) located in various organs including the heart, and triggering a pro-fibrotic signalling cascade [39][40][41]. ACE is also distributed throughout the cardiovascular system and renal tissues, allowing the local synthesis of Ang II [37][42].
1.4. Intracellular or Intracrine RAS
The pro-remodelling arm of the RAS may also be mediated by the intracellular synthesis of renin and Ang II in cardiac fibroblasts [43][44][45]. High glucose levels and isoproterenol elevate intracellular renin synthesis, which is converted to Ang II through intracellular ACE, subsequently elevating pro-fibrotic transforming growth factor-β (TGF-β) and collagen-1 synthesis in neonatal rat ventricular fibroblasts [46]. High-glucose-induced intracellular Ang II synthesis as well as superoxide production and cardiac fibrosis were only fully blocked by aliskiren (direct renin inhibitor) but not by ARB or ACE inhibitors [47].
2. microRNAs in the Regulation of RAS-Induced Cardiac Remodelling
MicroRNAs (miRNAs) are noncoding regulatory RNAs of ~22 nucleotides in length, involved in the silencing of genetic transcripts by binding to 3′ UTR of its target genes in eukaryotic cells [48][49]. A single miRNA can target a multitude of genes, while a single gene can be the target of a multitude of miRNAs [50]. While under physiological conditions, the main functions of miRNAs are considered to be the fine-tuning of gene expression; its silencing effect becomes more prominent under pathological conditions [51]. Since its relatively recent discovery, miRNA involvement has been implicated in the majority of pathological states, including RAS-induced cardiac remodelling [52][53][54][55][56]. As the following sections illustrate, miRNAs seem to directly target the RAS and Ang II signalling at all levels, including enzymes that are involved in the generation of Ang II (renin, ACE), its receptors (AT1R, AT2R), and compounds of Ang II signalling pathways.
2.1. miRNAs Involved in Ang II-Induced Cardiac Hypertrophy
Ang II-induced hypertrophy in cardiomyocytes is primarily mediated by AT1R through the canonical pathway, the epidermal growth factor receptor (EGFR) transactivation pathway, and the inflammatory pathway (reviewed in [57]).
The canonical pathway is initiated through the activation of the Gα13 protein, which in turn activates the calcineurin/NFAT, ERK1/2 pathways and RhoA [58][59]. RhoA is a small GTPase that induces further downstream activation of myocardin-related transcription factors, leading to the expression of both hypertrophic and fibrotic genes [60].
EGFR transactivation is initiated by the Ang II-induced increase in the intracellular Ca2+ and ROS, followed by the Src phosphorylation of ADAM metallopeptidase domain 17 (ADAM17). ADAM17 catalyses the shedding of the heparin-binding EGF-like growth factor (HB-EGF), the ligand for EGFR, resulting in a pro-hypertrophic MAPK, and phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) signalling [61].
Lastly, additional pro-hypertrophic growth-promoting signals can also be obtained from the AT1R-induced inflammatory pathway, which is mediated by the release of a variety of inflammatory cytokines including IL-1, IL-6, and TNF-α, along with NOX2 and NOX4 activation and subsequent ROS generation [62][63][64][65][66][67][68][69][70][71].
Furthermore, AT1R-induced mitochondrial NOX4 activation leads to elevated mitochondrial ROS, which induces hypertrophy-promoting autophagy [72]. Indeed, reduced ROS due to the cardioprotective Ang (1-7)/MasR axis reduces cardiac autophagy as well as hypertrophy, while the opposite action was observed with the deletion of the anti-inflammatory cytokine IL-10 [73][74].
The miRNA expression in Ang II-mediated cardiac hypertrophy is known to be deleterious, and now a growing list of miRNAs is associated with cardiac hypertrophy. Specifically, miR-155, miR-208, miR-132, miR-212, miR-21, miR-410, miR-495, miR-19a, miR-19b, and miR-20b have been associated with promoting hypertrophy, while miR-21-3p, miR-26a, miR-16, miR-98, miR-30a, 34a, miR-133a, miR-1, miR-99a, miR-101, and miR-129-3p have been associated with the prevention or reversal of cardiac hypertrophic growth; however, conflicting evidence also exists and is addressed in the following sections (Figure 2).
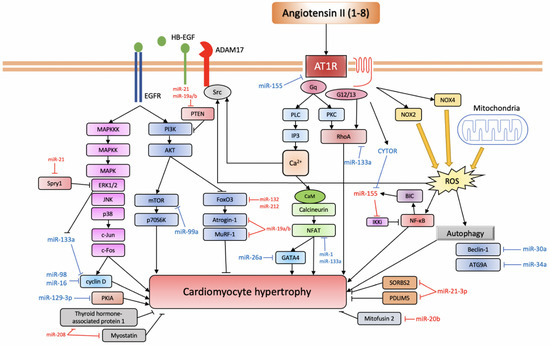
Figure 2. miRNAs involved in Ang II-induced cardiac hypertrophy.
AT1R triggers a variety of signalling pathways that can be divided into the canonical pathway depicted at the centre of the figure: the EGFR transactivation pathway depicted to the left and the inflammatory pathway depicted on the right (based on [57]). Anti-hypertrophic miRNAs are depicted in blue, while pro-hypertrophic ones are depicted in red.
AT1R, angiotensin II receptor type 1; PLC, phospholipase C; IP3, inositol triphosphate; PKC, protein kinase C; CaM, calmodulin; NFAT, nuclear factor of activated T-cell; CYTOR, cytoskeleton regulator RNA; BIC, B-cell integration cluster; IKKi, I-kappa-B kinase epsilon; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOX, NADPH oxidase; ROS, reactive oxygen species; ADAM17, ADAM metallopeptidase domain 17; HB-EGF, heparin-binding EGF-like growth factor; EGFR, epidermal growth factor receptor; MAPKKK, mitogen-activated protein kinase kinase kinase; MAPKK, mitogen-activated protein kinase kinase; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase 1/2; JNK, c-Jun N-terminal kinase; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; mTOR, mechanistic target of rapamycin; p70S6K, ribosomal protein S6 kinase 1; FoxO3, forkhead box protein O3; MuRF-1, muscle RING-finger protein-1; ATG9A, autophagy-related protein 9A; SORBS2, SH3 domain-containing protein 2; PDLIM5, PDZ and LIM domain 5; PKIA, protein kinase A inhibitor.
2.2. miRNAs in Ang II-Induced Cardiac Fibrosis
Ang II-induced ROS production plays a central role in the pro-fibrotic downstream signalling to AT1R. The primary sources of ROS are likely to be NOX2 and NOX4, which are the dominant isoforms in the heart, as well as the mitochondria [71][75][76]. Specifically, the mitochondrially located pattern recognition receptor nucleotide-binding domain and leucine-rich repeat-containing PYD-3 (NLRP3) represent a major source of Ang II- and TGF-β-induced ROS in mice cardiac fibroblasts, promoting fibrosis [77].
Although a substantial amount of ROS can oxidize biomolecules and directly contribute to cell damage and death, the transient increase in ROS triggered by growth factors, hormones, or various inflammatory cytokines can contribute to redox signalling and activate the stress response pathway [78]. In Ang II-treated cardiomyocytes and cardiac fibroblasts, ROS can activate a variety of signalling cascades, including AKT/mTOR, ERK1/2, p38, NF-κB, RhoA, matrix metalloproteinase-2 (MMP-2), and the TGF-β pathway that lead to the expression of fibrotic genes, such as extracellular matrix (ECM) proteins, MMP-9, α-smooth muscle actin (α-SMA), and periostin [71][79][80][81][82][83][84]. It is worth noting that the activation of each pathway possesses some redundancy, and intracellular Ca2+ and PKC-δ can also activate such pro-fibrotic ERK1/2 pathway, while ERK1/2 or p38 themselves can also augment the TGF-β pathway [85][86][87]. RhoA is activated downstream from NOX-dependent ROS generation, and by Gα13 and Gq/11 downstream from AT1R [60][84][88][89]. RhoA subsequently increases SRF activity and CTGF secretion, contributing to increased ECM deposits [89][90]. Moreover, in cardiac fibroblasts, NOX4-induced ROS downstream from AT1R leads to pro-inflammatory IL-18 expression, as well as MMP-9, lipoxygenase (LOX), and collagen expression, which in general favours fibroblast migration and ECM deposition [91]. ROS contributes to the TGF-β pathway, increasing the activity of both canonical SMAD-dependent pathways and noncanonical signalling through p38 and ERK1/2 signalling and SRF/CREB transcription factors [57][92]. Furthermore, as with hypertrophy, the transactivation of EGFR through ROS in cardiomyocytes also contributes to fibrosis via MAPK/ERK or PI3K/AKT pathways [93][94]. In addition to these redox-sensitive signalling proteins, ROS induces the cytoplasmic translocation of the RNA-binding protein HuR, which is followed by an augmented TGF-β pathway [95].
The multifactorial cytokine TGF-β family is a key regulator of pro-fibrotic genes, favouring ECM deposition in many organs including the heart [96]. It consists of three isoforms, of which TGF-β1 is mainly implicated in cardiac remodelling [97][98][99][100]. Extensive evidence suggests that Ang II can induce TGF-β1 expression through AT1R signalling, leading to auto-/para-crine responses in cardiomyocytes and cardiac fibroblasts [101]. In fact, the hypertrophic and fibrotic effect of Ang II is completely absent if TGF-β1 is knocked out [102]. In ventricular myocytes, upon Ang II binding to the Gq-coupled receptor AT1R, the NOX-induced sequential activation of PKC, MAPK, and AP-1 triggers TGF-β1 expression [103]. In VSMCs and primary rat cardiac fibroblasts, Ang II can also activate the SMAD pathway independent of TGF-β [104][105][106][107][108].
TGF-β1 signalling is initiated upon TGF-β binding to and assembly of TGF-β receptor type I and type II serine/threonine kinases (TGFBR1, TGFBR2), followed by the canonical SMAD pathway or the noncanonical non-SMAD pathways including Rho-like GTPase pathways, MAPK pathways, and PI3K/AKT pathways [109][110]. SMAD proteins can be categorized into three groups based on their functions [111]. The receptor-regulated R-SMADs (SMAD2, SMAD3) are substrates for TGF-β family receptors. The common Co-SMAD (SMAD4) functions as a common binding target for the R-SMADs and the complex can subsequently be transported into the nucleus. The inhibitory I-SMADs (SMAD6, SMAD7) compete with SMAD4 for R-SMAD binding, and upon binding, they inhibit R-SMAD function. The TGF-β/SMAD pathway has been implicated in inflammation and tissue injury-induced cardiac fibrosis [100]. Specifically, SMAD3 is particularly important for its pro-fibrotic effect, while inhibitory SMAD7 antagonizes it [112][113]. Curiously, SMAD2 deletion would increase fibrosis by upregulating SMAD3 [114]. Non-SMAD pathways in cardiac fibrosis have yet to be systematically studied, but at least TGF-β-activated kinase 1 (TAK1) has been suggested to play a role in the process through its action on MKKs and the resulting phosphorylation of JNK and p38 [115][116][117]. The level of TAK1 activity was found to be elevated 7 days after increased mechanical load with aortic banding, while an activating mutation induces cardiac hypertrophy and interstitial fibrosis [116].
Several studies have demonstrated the involvement of miRNAs in regulating myocardial fibrosis in the settings of myocardial ischemia or mechanical overload. Namely, miR-21, miR-433, miR-503, miR-34a, and miR-155 have been shown to favour fibrogenesis, being pro-fibrotic miRNAs, while miR-26a, miR133a, miR-19a/b-3p, miR-29b, miR-22, and miR-let-7i have been found to be anti-fibrotic miRNAs (Figure 3).
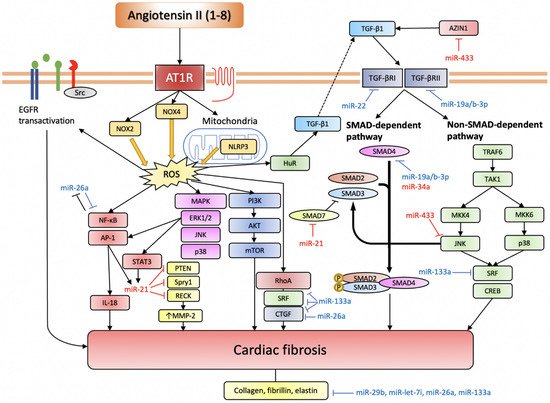
Figure 3. miRNAs involved in Ang II-induced cardiac fibrosis.
Ang II induces ROS production through NOX2, NOX4, and NLRP3. Intracellular ROS serves an important signalling function by activating various pro-fibrotic pathways (based on [57]). TGF-β pathway is another indispensable pathway for Ang II-induced cardiac fibrosis, and it can also be activated through elevated ROS. The EGFR transactivation pathway is more important in cardiomyocytes. Anti-fibrotic miRNAs are depicted in blue, while pro-fibrotic miRNAs are depicted in red.
AT1R, angiotensin II receptor type 1; NOX, NADPH oxidase; NLRP3, nucleotide-binding domain and leucine-rich repeat-containing PYD-3; ROS, reactive oxygen species; EGFR, epidermal growth factor receptor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; AP-1, activator protein-1; IL-18, interleukin-18; STAT3, signal transducer and activator of transcription 3; PTEN, phosphatase and tensin homolog; Spry1, sprouty homolog 1; RECK, reversion-inducing cysteine-rich protein with Kazal motifs; MMP-2, matrix metalloproteinase-2; MAPK, mitogen-activated protein kinase; ERK1/2, extracellular signal-regulated kinase 1/2; JNK, c-Jun N-terminal kinase; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B; mTOR, mechanistic target of rapamycin; HuR, human antigen R; TGF-β, transforming growth factor-β; SMAD, small mothers against decapentaplegic homolog; TRAF6, TNF receptor associated factor-6; TAK1, TGF-β-activated kinase 1; MKK, mitogen-activated protein kinase kinase; SRF, serum response factor; CREB, cAMP response element-binding protein.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094762
References
- Goodman, L.S.; Brunton, L.L.; Chabner, B.; Knollmann, B.C. Goodman & Gilman’s Pharmacological Basis of Therapeutics; McGraw-Hill: New York, NY, USA, 2011; pp. 721–741.
- Von Lueder, T.G.; Krum, H. RAAS inhibitors and cardiovascular protection in large scale trials. Cardiovasc. Drugs Ther. 2013, 27, 171–179.
- Simko, F.; Pechanova, O.; Repova-Bednarova, K.; Krajcirovicova, K.; Celec, P.; Kamodyova, N.; Zorad, S.; Kucharska, J.; Gvozdjakova, A.; Adamcova, M.; et al. Hypertension and cardiovascular remodelling in rats exposed to continuous light: Protection by ACE-inhibition and melatonin. Mediators Inflamm. 2014, 2014, 703175:1–703175:10.
- Simko, F.; Pechanova, O.; Krajcirovicova, K.; Matuskova, J.; Pelouch, V.; Adamcova, M.; Paulis, L. Effects of captopril, spironolactone, and simvastatin on the cardiovascular system of non-diseased Wistar rats. Int. J. Cardiol. 2015, 190, 128–130.
- Simko, F.; Pechanova, O.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Celec, P.; Tothova, L.; Vrankova, S.; Balazova, L.; Zorad, S.; et al. Lactacystin-Induced Model of Hypertension in Rats: Effects of Melatonin and Captopril. Int. J. Mol. Sci. 2017, 18, 1612.
- Simko, F.; Baka, T.; Poglitsch, M.; Repova, K.; Aziriova, S.; Krajcirovicova, K.; Zorad, S.; Adamcova, M.; Paulis, L. Effect of Ivabradine on a Hypertensive Heart and the Renin-Angiotensin-Aldosterone System in L-NAME-Induced Hypertension. Int. J. Mol. Sci. 2018, 19, 3017.
- Simko, F.; Hrenak, J.; Adamcova, M.; Paulis, L. Renin-Angiotensin-Aldosterone System: Friend or Foe—The Matter of Balance. Insight on History, Therapeutic Implications and COVID-19 Interactions. Int. J. Mol. Sci. 2021, 22, 3217.
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J. Cell. Signal. 2016, 1, 111.
- Ruiz-Ortega, M.; Lorenzo, O.; Rupérez, M.; König, S.; Wittig, B.; Egido, J. Angiotensin II activates nuclear transcription factor kappaB through AT(1) and AT(2) in vascular smooth muscle cells: Molecular mechanisms. Circ. Res. 2000, 86, 1266–1272.
- Schieffer, B.; Wirger, A.; Meybrunn, M.; Seitz, S.; Holtz, J.; Riede, U.N.; Drexler, H. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation 1994, 89, 2273–2282.
- Sadoshima, J.; Izumo, S. Molecular characterization of angiotensin II--induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ. Res. 1993, 73, 413–423.
- Simko, F.; Pechanova, O.; Pelouch, V.; Krajcirovicova, K.; Mullerova, M.; Bednarova, K.; Adamcova, M.; Paulis, L. Effect of melatonin, captopril, spironolactone and simvastatin on blood pressure and left ventricular remodelling in spontaneously hypertensive rats. J. Hypertens. Suppl. 2009, 27, S5–S10.
- AbdAlla, S.; Lother, H.; Abdel-tawab, A.M.; Quitterer, U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J. Biol. Chem. 2001, 276, 39721–39726.
- Uehara, Y.; Miura, S.; Yahiro, E.; Saku, K. Non-ACE pathway-induced angiotensin II production. Curr. Pharm. Des. 2013, 19, 3054–3059.
- Simko, F.; Simko, J.; Fabryová, M. ACE-inhibition and angiotensin II receptor blockers in chronic heart failure: Pathophysiological consideration of the unresolved battle. Cardiovasc. Drugs Ther. 2003, 17, 287–290.
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9.
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243.
- Hrenak, J.; Simko, F. Renin-Angiotensin System: An Important Player in the Pathogenesis of Acute Respiratory Distress Syndrome. Int. J. Mol. Sci. 2020, 21, E8038.
- Ferrario, C.M.; Chappell, M.C.; Tallant, E.A.; Brosnihan, K.B.; Diz, D.I. Counterregulatory actions of angiotensin-(1-7). Hypertension 1997, 30, 535–541.
- Huentelman, M.J.; Grobe, J.L.; Vazquez, J.; Stewart, J.M.; Mecca, A.P.; Katovich, M.J.; Ferrario, C.M.; Raizada, M.K. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp. Physiol. 2005, 90, 783–790.
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828.
- Santos, R.A.; Ferreira, A.J.; Simões E Silva, A.C. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp. Physiol. 2008, 93, 519–527.
- Ferrario, C.M. Angiotensin-converting enzyme 2 and angiotensin-(1-7): An evolving story in cardiovascular regulation. Hypertension 2006, 47, 515–521.
- Schindler, C.; Bramlage, P.; Kirch, W.; Ferrario, C.M. Role of the vasodilator peptide angiotensin-(1-7) in cardiovascular drug therapy. Vasc. Health Risk Manag. 2007, 3, 125–137.
- Walters, P.E.; Gaspari, T.A.; Widdop, R.E. Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension 2005, 45, 960–966.
- Tetzner, A.; Gebolys, K.; Meinert, C.; Klein, S.; Uhlich, A.; Trebicka, J.; Villacañas, Ó.; Walther, T. G-Protein-Coupled Receptor MrgD Is a Receptor for Angiotensin-(1-7) Involving Adenylyl Cyclase, cAMP, and Phosphokinase A. Hypertension 2016, 68, 185–194.
- Schleifenbaum, J. Alamandine and Its Receptor MrgD Pair Up to Join the Protective Arm of the Renin-Angiotensin System. Front. Med. (Lausanne) 2019, 6, 107.
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129.
- Jankowski, V.; Vanholder, R.; van der Giet, M.; Tölle, M.; Karadogan, S.; Gobom, J.; Furkert, J.; Oksche, A.; Krause, E.; Tran, T.N.; et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 297–302.
- Jankowski, V.; Tölle, M.; Santos, R.A.; Günthner, T.; Krause, E.; Beyermann, M.; Welker, P.; Bader, M.; Pinheiro, S.V.; Sampaio, W.O.; et al. Angioprotectin: An angiotensin II-like peptide causing vasodilatory effects. FASEB J. 2011, 25, 2987–2995.
- Albiston, A.L.; McDowall, S.G.; Matsacos, D.; Sim, P.; Clune, E.; Mustafa, T.; Lee, J.; Mendelsohn, F.A.; Simpson, R.J.; Connolly, L.M.; et al. Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin-regulated aminopeptidase. J. Biol. Chem. 2001, 276, 48623–48626.
- Wright, J.W.; Miller-Wing, A.V.; Shaffer, M.J.; Higginson, C.; Wright, D.E.; Hanesworth, J.M.; Harding, J.W. Angiotensin II(3-8) (ANG IV) hippocampal binding: Potential role in the facilitation of memory. Brain Res. Bull. 1993, 32, 497–502.
- Hrenak, J.; Paulis, L.; Simko, F. N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP): Potential target molecule in research of heart, kidney and brain. Curr. Pharm. Des. 2015, 21, 5135–5143.
- Chen, L.J.; Xu, R.; Yu, H.M.; Chang, Q.; Zhong, J.C. The ACE2/Apelin Signaling, MicroRNAs, and Hypertension. Int. J. Hypertens. 2015, 2015, 896861.
- Yu, X.H.; Tang, Z.B.; Liu, L.J.; Qian, H.; Tang, S.L.; Zhang, D.W.; Tian, G.P.; Tang, C.K. Apelin and its receptor APJ in cardiovascular diseases. Clin. Chim. Acta 2014, 428, 1–8.
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The Role of Apelin in Cardiovascular Diseases, Obesity and Cancer. Front. Physiol. 2018, 9, 557.
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803.
- Simko, F.; Simko, J. Heart failure and angiotensin converting enzyme inhibition: Problems and perspectives. Physiol. Res. 1999, 48, 1–8.
- Nguyen, G.; Danser, A.H. Prorenin and (pro)renin receptor: A review of available data from in vitro studies and experimental models in rodents. Exp. Physiol. 2008, 93, 557–563.
- Mahmud, H.; Silljé, H.H.; Cannon, M.V.; van Gilst, W.H.; de Boer, R.A. Regulation of the (pro)renin-renin receptor in cardiac remodelling. J. Cell Mol. Med. 2012, 16, 722–729.
- Ichihara, A.; Yatabe, M.S. The (pro)renin receptor in health and disease. Nat. Rev. Nephrol. 2019, 15, 693–712.
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An Update on the Tissue Renin Angiotensin System and Its Role in Physiology and Pathology. J. Cardiovasc. Dev. Dis. 2019, 6, E14.
- Singh, V.P.; Le, B.; Bhat, V.B.; Baker, K.M.; Kumar, R. High-glucose-induced regulation of intracellular ANG II synthesis and nuclear redistribution in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H939–H948.
- Kumar, R.; Singh, V.P.; Baker, K.M. The intracellular renin-angiotensin system: A new paradigm. Trends Endocrinol. Metab. 2007, 18, 208–214.
- Kumar, R.; Singh, V.P.; Baker, K.M. The intracellular renin-angiotensin system in the heart. Curr. Hypertens. Rep. 2009, 11, 104–110.
- Singh, V.P.; Baker, K.M.; Kumar, R. Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: Role in extracellular matrix production. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1675–H1684.
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57, 3297–3306.
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108.
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524.
- Doench, J.G.; Petersen, C.P.; Sharp, P.A. siRNAs can function as miRNAs. Genes Dev. 2003, 17, 438–442.
- Van Rooij, E. The art of microRNA research. Circ. Res. 2011, 108, 219–234.
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478.
- Chen, C.; Ponnusamy, M.; Liu, C.; Gao, J.; Wang, K.; Li, P. MicroRNA as a Therapeutic Target in Cardiac Remodeling. Biomed. Res. Int. 2017, 2017, 1278436.
- Deiuliis, J.; Mihai, G.; Zhang, J.; Taslim, C.; Varghese, J.J.; Maiseyeu, A.; Huang, K.; Rajagopalan, S. Renin-sensitive microRNAs correlate with atherosclerosis plaque progression. J. Hum. Hypertens. 2014, 28, 251–258.
- Butterworth, M.B. Role of microRNAs in aldosterone signaling. Curr. Opin. Nephrol. Hypertens. 2018, 27, 390–394.
- Butterworth, M.B. Non-coding RNAs and the mineralocorticoid receptor in the kidney. Mol. Cell Endocrinol. 2021, 521, 111115.
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738.
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262.
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438.
- Takefuji, M.; Wirth, A.; Lukasova, M.; Takefuji, S.; Boettger, T.; Braun, T.; Althoff, T.; Offermanns, S.; Wettschureck, N. G(13)-mediated signaling pathway is required for pressure overload-induced cardiac remodeling and heart failure. Circulation 2012, 126, 1972–1982.
- Forrester, S.J.; Kawai, T.; O’Brien, S.; Thomas, W.; Harris, R.C.; Eguchi, S. Epidermal Growth Factor Receptor Transactivation: Mechanisms, Pathophysiology, and Potential Therapies in the Cardiovascular System. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 627–653.
- Jia, L.; Li, Y.; Xiao, C.; Du, J. Angiotensin II induces inflammation leading to cardiac remodeling. Front. Biosci. (Landmark Ed.) 2012, 17, 221–231.
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257.
- Funakoshi, Y.; Ichiki, T.; Ito, K.; Takeshita, A. Induction of interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension 1999, 34, 118–125.
- Zhang, W.; Wang, W.; Yu, H.; Zhang, Y.; Dai, Y.; Ning, C.; Tao, L.; Sun, H.; Kellems, R.E.; Blackburn, M.R.; et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension 2012, 59, 136–144.
- Luther, J.M.; Gainer, J.V.; Murphey, L.J.; Yu, C.; Vaughan, D.E.; Morrow, J.D.; Brown, N.J. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 2006, 48, 1050–1057.
- Sriramula, S.; Francis, J. Tumor Necrosis Factor-Alpha Is Essential for Angiotensin II-Induced Ventricular Remodeling: Role for Oxidative Stress. PLoS ONE 2015, 10, e0138372.
- Dandona, P.; Dhindsa, S.; Ghanim, H.; Chaudhuri, A. Angiotensin II and inflammation: The effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J. Hum. Hypertens. 2007, 21, 20–27.
- Bendall, J.K.; Cave, A.C.; Heymes, C.; Gall, N.; Shah, A.M. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation 2002, 105, 293–296.
- Zhang, M.; Prosser, B.L.; Bamboye, M.A.; Gondim, A.N.S.; Santos, C.X.; Martin, D.; Ghigo, A.; Perino, A.; Brewer, A.C.; Ward, C.W.; et al. Contractile Function During Angiotensin-II Activation: Increased Nox2 Activity Modulates Cardiac Calcium Handling via Phospholamban Phosphorylation. J. Am. Coll. Cardiol. 2015, 66, 261–272.
- Zhao, Q.D.; Viswanadhapalli, S.; Williams, P.; Shi, Q.; Tan, C.; Yi, X.; Bhandari, B.; Abboud, H.E. NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation 2015, 131, 643–655.
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ. Res. 2011, 108, 837–846.
- Lin, L.; Liu, X.; Xu, J.; Weng, L.; Ren, J.; Ge, J.; Zou, Y. Mas receptor mediates cardioprotection of angiotensin-(1-7) against Angiotensin II-induced cardiomyocyte autophagy and cardiac remodelling through inhibition of oxidative stress. J. Cell. Mol. Med. 2016, 20, 48–57.
- Kishore, R.; Krishnamurthy, P.; Garikipati, V.N.; Benedict, C.; Nickoloff, E.; Khan, M.; Johnson, J.; Gumpert, A.M.; Koch, W.J.; Verma, S.K. Interleukin-10 inhibits chronic angiotensin II-induced pathological autophagy. J. Mol. Cell. Cardiol. 2015, 89, 203–213.
- Wen, H.; Gwathmey, J.K.; Xie, L.H. Oxidative stress-mediated effects of angiotensin II in the cardiovascular system. World J. Hypertens. 2012, 2, 34–44.
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J 2006, 20, 1546–1548.
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R.; Fedak, P.W.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 289, 19571–19584.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390.
- Sag, C.M.; Santos, C.X.; Shah, A.M. Redox regulation of cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 73, 103–111.
- Belo, V.A.; Guimarães, D.A.; Castro, M.M. Matrix Metalloproteinase 2 as a Potential Mediator of Vascular Smooth Muscle Cell Migration and Chronic Vascular Remodeling in Hypertension. J. Vasc. Res. 2015, 52, 221–231.
- Luchtefeld, M.; Grote, K.; Grothusen, C.; Bley, S.; Bandlow, N.; Selle, T.; Strüber, M.; Haverich, A.; Bavendiek, U.; Drexler, H.; et al. Angiotensin II induces MMP-2 in a p47phox-dependent manner. Biochem. Biophys. Res. Commun. 2005, 328, 183–188.
- Li, L.; Fan, D.; Wang, C.; Wang, J.Y.; Cui, X.B.; Wu, D.; Zhou, Y.; Wu, L.L. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac fibroblasts. Cardiovasc. Res. 2011, 91, 80–89.
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120.
- Olson, E.R.; Shamhart, P.E.; Naugle, J.E.; Meszaros, J.G. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cdelta and intracellular calcium in adult rat cardiac fibroblasts. Hypertension 2008, 51, 704–711.
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18.
- Omura, T.; Yoshiyama, M.; Kim, S.; Matsumoto, R.; Nakamura, Y.; Izumi, Y.; Ichijo, H.; Sudo, T.; Akioka, K.; Iwao, H.; et al. Involvement of apoptosis signal-regulating kinase-1 on angiotensin II-induced monocyte chemoattractant protein-1 expression. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 270–275.
- Balakumar, P.; Jagadeesh, G. A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell. Signal. 2014, 26, 2147–2160.
- Ongherth, A.; Pasch, S.; Wuertz, C.M.; Nowak, K.; Kittana, N.; Weis, C.A.; Jatho, A.; Vettel, C.; Tiburcy, M.; Toischer, K.; et al. p63RhoGEF regulates auto- and paracrine signaling in cardiac fibroblasts. J. Mol. Cell. Cardiol. 2015, 88, 39–54.
- Li, C.; Zhen, G.; Chai, Y.; Xie, L.; Crane, J.L.; Farber, E.; Farber, C.R.; Luo, X.; Gao, P.; Cao, X.; et al. RhoA determines lineage fate of mesenchymal stem cells by modulating CTGF-VEGF complex in extracellular matrix. Nat. Commun. 2016, 7, 11455.
- Somanna, N.K.; Valente, A.J.; Krenz, M.; Fay, W.P.; Delafontaine, P.; Chandrasekar, B. The Nox1/4 Dual Inhibitor GKT137831 or Nox4 Knockdown Inhibits Angiotensin-II-Induced Adult Mouse Cardiac Fibroblast Proliferation and Migration. AT1 Physically Associates With Nox4. J. Cell Physiol. 2016, 231, 1130–1141.
- Liu, R.M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577.
- Moriguchi, Y.; Matsubara, H.; Mori, Y.; Murasawa, S.; Masaki, H.; Maruyama, K.; Tsutsumi, Y.; Shibasaki, Y.; Tanaka, Y.; Nakajima, T.; et al. Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-beta synthesis via transcriptional and posttranscriptional mechanisms. Circ. Res. 1999, 84, 1073–1084.
- Peng, K.; Tian, X.; Qian, Y.; Skibba, M.; Zou, C.; Liu, Z.; Wang, J.; Xu, Z.; Li, X.; Liang, G. Novel EGFR inhibitors attenuate cardiac hypertrophy induced by angiotensin II. J. Cell. Mol. Med. 2016, 20, 482–494.
- Bai, D.; Ge, L.; Gao, Y.; Lu, X.; Wang, H.; Yang, G. Cytoplasmic translocation of HuR contributes to angiotensin II induced cardiac fibrosis. Biochem. Biophys. Res. Commun. 2015, 463, 1273–1277.
- Border, W.A.; Noble, N.A. Transforming growth factor beta in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292.
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor Beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 2015, 137823.
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 2000, 71, 418–435.
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195.
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783.
- Rosenkranz, S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432.
- Schultz, J.E.J.; Witt, S.A.; Glascock, B.J.; Nieman, M.L.; Reiser, P.J.; Nix, S.L.; Kimball, T.R.; Doetschman, T. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J. Clin. Investig. 2002, 109, 787–796.
- Wenzel, S.; Taimor, G.; Piper, H.M.; Schlüter, K.D. Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-beta expression in adult ventricular cardiomyocytes. FASEB J. 2001, 15, 2291–2293.
- Rodríguez-Vita, J.; Sánchez-López, E.; Esteban, V.; Rupérez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517.
- Ruiz-Ortega, M.; Rodríguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206.
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ. Res. 2006, 98, 1032–1039.
- Hao, J.; Wang, B.; Jones, S.C.; Jassal, D.S.; Dixon, I.M. Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H3020–H3030.
- Carvajal, G.; Rodríguez-Vita, J.; Rodrigues-Díez, R.; Sánchez-López, E.; Rupérez, M.; Cartier, C.; Esteban, V.; Ortiz, A.; Egido, J.; Mezzano, S.A.; et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int. 2008, 74, 585–595.
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700.
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139.
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810.
- Li, J.H.; Zhu, H.J.; Huang, X.R.; Lai, K.N.; Johnson, R.J.; Lan, H.Y. Smad7 inhibits fibrotic effect of TGF-Beta on renal tubular epithelial cells by blocking Smad2 activation. J. Am. Soc. Nephrol. 2002, 13, 1464–1472.
- Wang, B.; Omar, A.; Angelovska, T.; Drobic, V.; Rattan, S.G.; Jones, S.C.; Dixon, I.M. Regulation of collagen synthesis by inhibitory Smad7 in cardiac myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1282–H1290.
- Meng, X.M.; Huang, X.R.; Chung, A.C.; Qin, W.; Shao, X.; Igarashi, P.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1477–1487.
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657.
- Zhang, D.; Gaussin, V.; Taffet, G.E.; Belaguli, N.S.; Yamada, M.; Schwartz, R.J.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D.; Schneider, M.D. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000, 6, 556–563.
- Wang, W.; Zhou, G.; Hu, M.C.; Yao, Z.; Tan, T.H. Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor beta (TGF-beta)-activated kinase (TAK1), a kinase mediator of TGF beta signal transduction. J. Biol. Chem. 1997, 272, 22771–22775.
This entry is offline, you can click here to edit this entry!