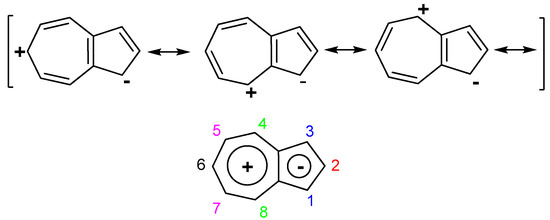
The nonalternant aromatic azulene, an isomer of alternant naphthalene, differs from the latter in peculiar properties. The large polarization of the π-electron system over the seven and five rings gives to azulene electrophile property a pronounced tendency to donate electrons to an acceptor, substituted at azulene 1 position. This paper presents cases in which azulene transfers electrons to a suitable acceptor as methylium ions, positive charged heteroaromatics and examples of neutral molecules that can accept electrons.
- methylium ion
- heteroaromatics
- π-electron transfer
- azulene
1. Introduction
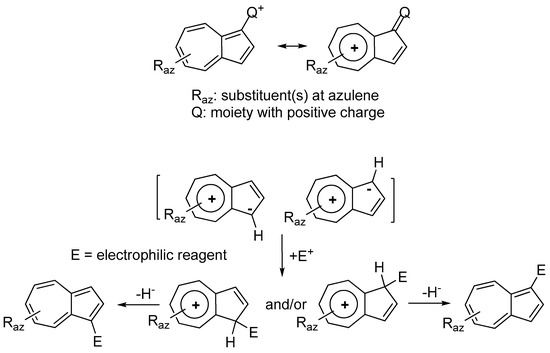
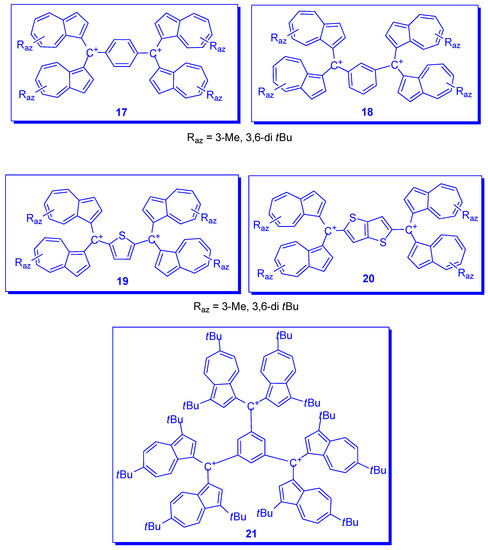
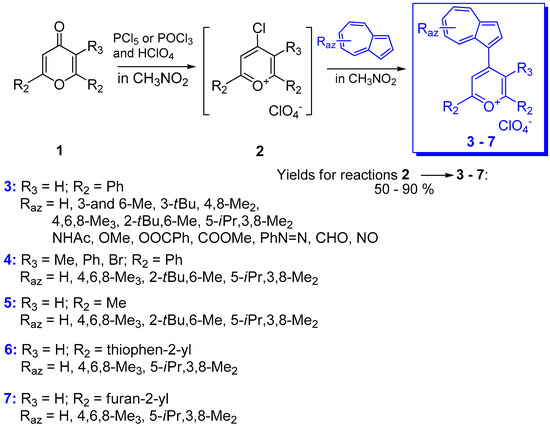
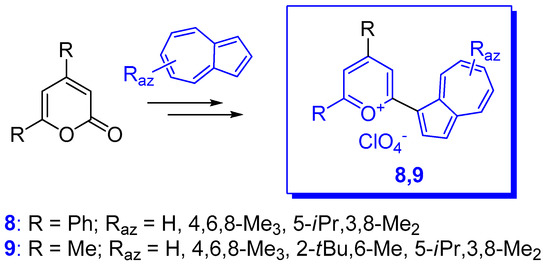
2. Triaryl-methylium Ion with at Least One Azulen-1-yl Moiety
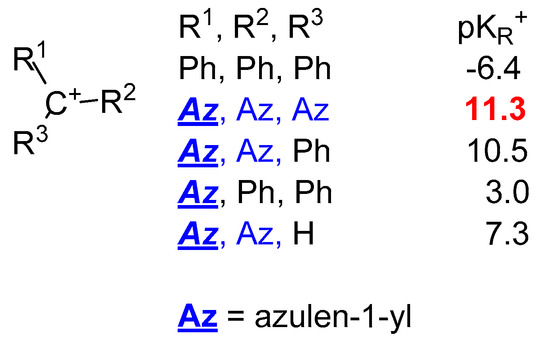
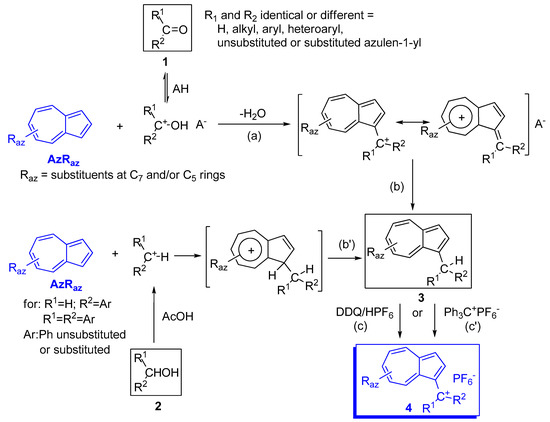
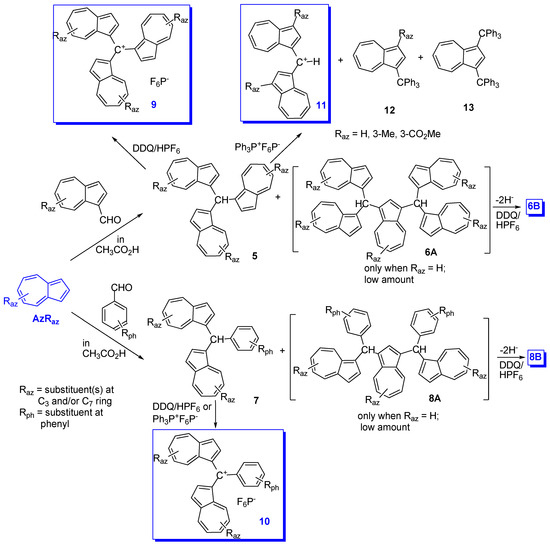
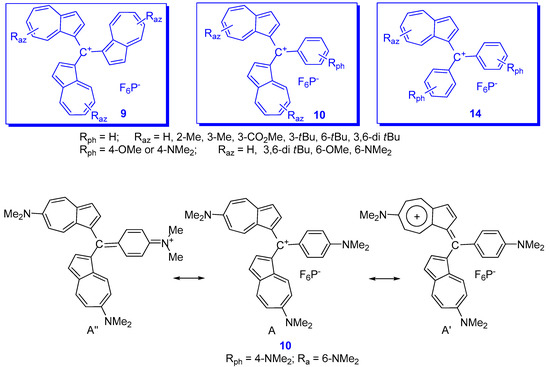
| Raz | Rph for 10 and 14 |
pKR+ | ||
|---|---|---|---|---|
| 9 | 10 | 14 | ||
| H | H | 11.3 | 10.5 | 3.0 |
| 3-Me | H | 11.4 | 10.8 | 3.7 |
| 3-tBu | - | 13.2 | - | - |
| 3,6-di tBu | H | 14.3 | 12.4 | 4.6 |
| 6-OMe | - | >14.0 | - | - |
| H | 4-OMe | - | 11.7 | - |
| 6-OMe | 4-OMe | - | >14.0 | - |
| 3,6-di tBu | 4-OMe | - | 13.4 | - |
| H | 4-Me2N | - | 13.2 | - |
| 3,6-di tBu | 4-Me2N | - | 13.8 | - |
| H | 4-Me2N | - | - | 12.6 |
| 3,6-di tBu | 4-Me2N | - | - | 13.3 |
| 6- Me2N | - | 24.3 | - | - |
| 6- Me2N | 4-Me2N | - | 21.5 | - |
| 6- Me2N | 4-Me2N | - | - | 14.0 |
| 3-Me (18a) | 2-thienyl | - | 11.2 | - |
| 3-tBu (18a) | 2-thienyl | - | 11.8 | - |
| 3-Me (18b) | 3-thienyl | - | 11.4 | - |
| 3- tBu (18b) | 3-thienyl | - | 12.4 | - |
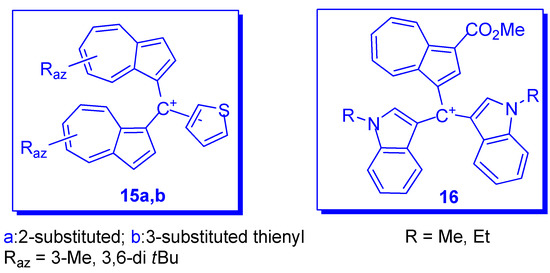
3. Charged Hetero-aryls Stabilized by Azulen-1-yl Moieties
| 1H | Azulene | 1-Phenyl- azulene |
4-(Azulen-1-yl)- 2,6-diphenylpyridine |
Salt 3 a | Salt 5 b | Salt 10 c |
|---|---|---|---|---|---|---|
| 3′/5′H | - | - | 7.95 | 8.83 | 8.06 | 8.32 |
| 2 | 7.81 | 8.02 | 8.18 | 8.92 | 8.45 | 8.18 |
| 3 | 7.30 | 7.43 | 7.53 | 7.77 | 7.51 | 7.35 |
| 4 | 8.23 | 8.34 | 8.44 | 8.89 | 8.65 | 8.44 |
| 5 | 7.05 | 7.14 | 7.28 | 8.03 | 7.77 | 7.28 |
| 6 | 7.45 | 7.58 | 7.45 | 8.31 | 8.07 | 7.72 |
| 7 | 7.05 | 7.14 | 7.29 | 8.14 | 7.80 | 7.29 |
| 8 | 8.23 | 8.55 | 8.71 | 9.57 | 9.12 | 8.71 |
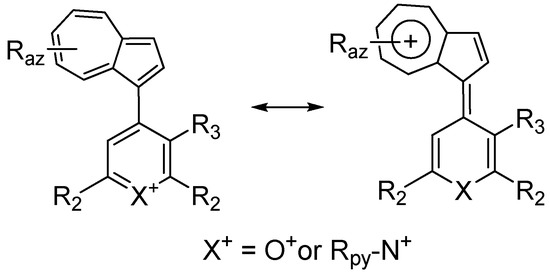
| Compd. * | Raz | R3 | Dihedral Angle (in °) [48] |
|---|---|---|---|
| 3 (R2 = Ph) | H | H | 24 |
| H | Me | 38 | |
| 2-tBu-6-Me | H | 50 | |
| 5 (R2 = Me) | H | H | 22 |
| 2-tBu-6-Me | H | 45 | |
| 10 (R2 = Me; Rpy = nBu) |
H | H | 29 |
| H | Me | 42 | |
| 2-tBu-6-Me | H | 54 | |
| 10 (R2 = Ph; Rpy = Me) | H | H | 39 |

- Other systems stabilized by azulen-1-yl moieties
In this last chapter reference will be made to some special cases of azulen-1-yl moiety participation to the stability of some carbocations and even of some neutral molecules.
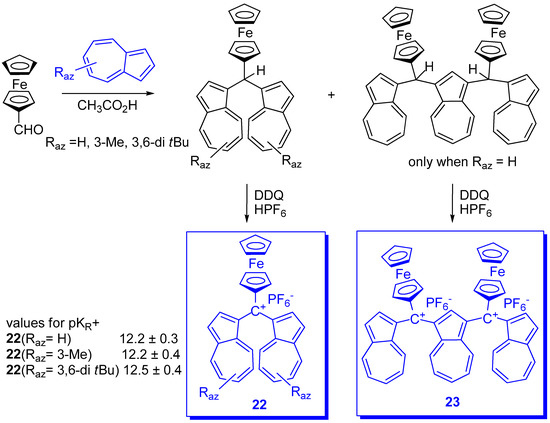
In connection with the stabilization of triarylmethylium ions by at least one azulen-1-yl moiety discussed before, the delocalized monocarbenium ions, salt 23, stabilized by largely conjugate π electrons including azulen-1-yl moieties next is presented. The salt 23 was obtained by condensation of all-trans-retinal with azulenes (Scheme 18) [50].
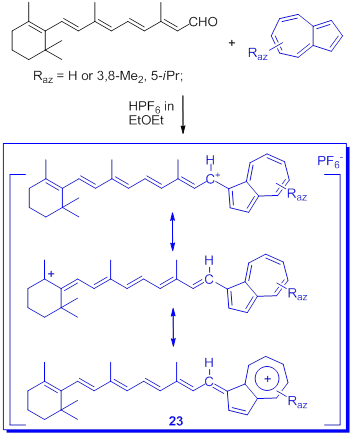
Scheme 18. Condensation of all-trans-retinal with azulenes.
Considering the good electron donor property of both ferrocene and of azulen-1-yls, Farrell et al. [51] used ferrocene-azulenium carbocations and their vinylogs as tetrafluoroborates to investigate a novel series of monometallic monocations, 24n. Starting from formyl ferrocene and azulenes as precursors, the yields of products 24n were between 58 % and 68 % (Scheme 19).
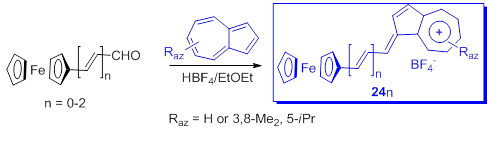
Scheme 19. Ferrocene-azulenium carbocations.
Contrary to the very low stability of most carbocations with hydrocarbon skeleton, some cyclic cations as cyclopropenyl [52] have remarkable stability. In fact, this system is the simplest aromatic Hückel system. The substitution of this cation with three cyclopropyl [53] or three guaiazulene moieties [54] dramatically increases the value of pKR+ compared with the compound with three phenyl at ring as it can be seen in Scheme 20. From the data in Scheme 20 it results the high difference between the pKR+ for triphenylmethylium and the tri(azulen-1-yl)methylium cation as compared with the difference between cyclopropenylium cations 25(R = Ph) and 25(R = guaiazulene-3-yl). The compound 25(R = guaiazulene-3-y) was obtained in the Friedel-Crafts reaction of C3Cl3+AlCl4 (prepared from tetrachlorocyclopropene and AlCl3) with 3 molar equivalents of guaiazulene in dichloromethane at -70 oC [54].
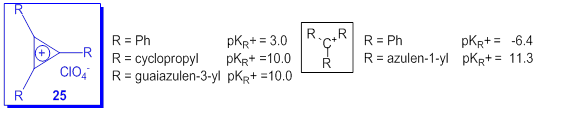
Scheme 20. pKR+ values for some carbocations with hydrocarbon skeleton.
Regarding the azulen-1-yl system contribution to the charge distribution in neutral molecules, without detailing this extensive subject that goes beyond the subject of this review, only some informative example will be presented below.
The azulenyl squarines, that will be discussed briefly, already synthesized in 1966 [55], have interested a number of researchers due to the optical properties of the compounds as well as for some biological applications. It should be mentioned that in the over 50 years since the birth of the first azulenyl squarines a huge number of compounds have been reported, some of them being covered by patents and in 2017 a consistent review was published on this topic [56]. The general procedure for obtaining these compounds, 26, is shown in Scheme 21 [57]. The presence of electron-donating groups in an aromatic system employed in a squaraine synthesis tends to favor the reaction and indeed the yield of reactions between azulene and squaric acid proceed satisfactorily.
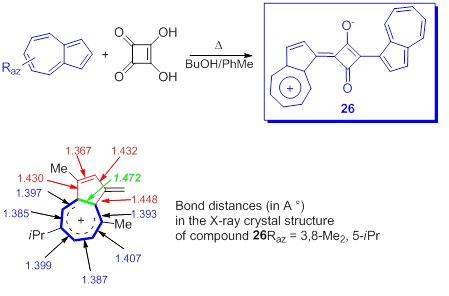
Scheme 21. Azulen-1-yl squarines.
Without going into details, some information on the structure of the azulenyl squarines is required. The X-ray crystal structure of compound 26(Raz = H or 3,8-Me2, 5-iPr) [56,58] shows the shorter C1-C2 bond as compared with the other five ring bonds. At the same time the bond length for six bonds in big ring are reduced as compared with the distance between the atoms in five membered ring and the longer azulene bond is between C3a and C8a. (Scheme 21). These observations seem to suggest a more efficient π–electron conjugation over C3a-C4-C5-C6-C7-C8-C8a than a tropylium structure.
The presence of [18]annulene substructures with 26π electrons within the macrocycle is responsible for the aromatic characteristics of porphyrins. According to this model, a large number of other compounds, including some containing azulene systems in the molecule, have been made over time. Lash et al. had the idea of replacing one or more pyrrole rings in the porphyrin macrocycle with the five ring of azulene so edifying a new class of compounds, azuliporphyrins and related carbaporphyrinoid compounds [58].
One of the synthesis of azuliporphyrins is based on the application of the “3+1”variant of the MacDonald reaction as it can be seen in Scheme 22 [58]. Azulene-1,3-dialdehyde was condensed with tripyrrane in the presence of HBF4, in dichloromethane followed by oxidation with DDQ. The compounds 27, porphyrin analogues, due to the participation of the five azulene ring at the stabilization of product, were generated in good yields. A large number of azuliporphyrins were also obtained by Lash's research team from which only two more examples, 28 and 29 were selected. In the last compound two azulenes moieties were inserted into the molecular structure.
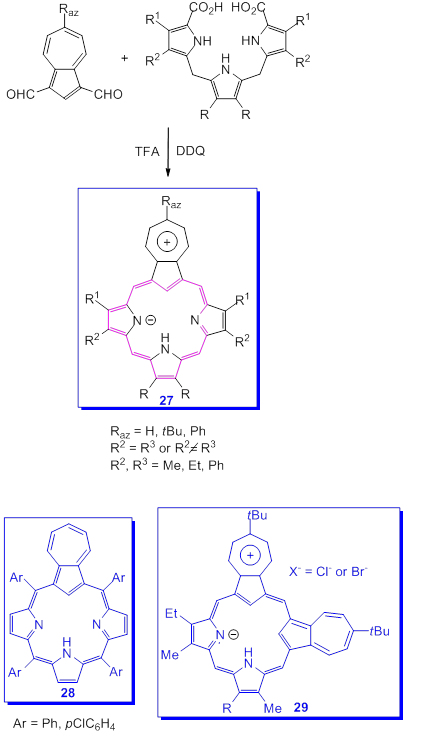
Scheme 22. Synthesis of azuliporphyrins.
Finally and as a curiosity, the attempts to realize a system analog to that of porphyrin, in which the four five-membered rings belong to azulenes [59,60] will be presented. Starting from theoretically exciting however presumed quatyrin system (Scheme 23), analog to that of porphyrin, the beginning was the preparation of calix[4]azulenes, 30, by reacting azulenes with paraformaldehyde in the presence of florisil. However the realized molecules did not have the desired structure namely [18]annulene substructures with 26π electrons. Using the tetraaryl compound 31, after oxidation with DDQ and an addition of HBF4·Et2O, the tetracation 32 was generated. Surprisingly, the chemical shifts in 1H-NMR spectrum of 32 are consistent with a non-aromatic structure. This finding as well as the lack of coplanarity of the macrocycle, determined by X-ray spectra, seems to indicate the predominant participation of the structure 32B with the charges located at the four Csp2 compared to the structure 32A with the tropylium ions. Thus, the macrocyclic aromatic ring current is essentially absent.
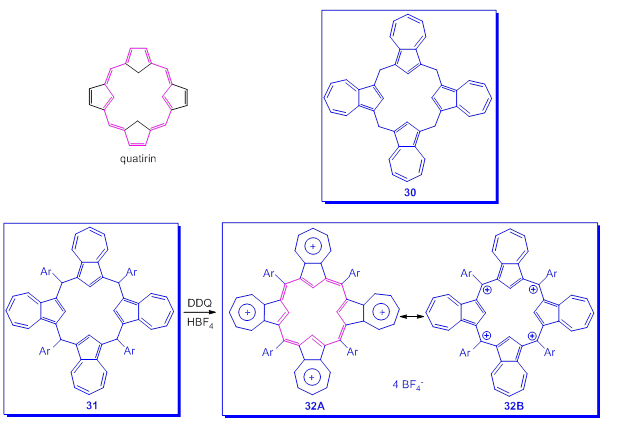
Scheme 23. Macrocycles including azulene moieties.
4. Conclusions
Azulenes, these exceptional compounds, related to natural derivatives, have a special structure that gives them properties totally different from those of alternating aromatic hydrocarbons and even from the nonalternating ones with which they are related. The paper proposes a brief look at one of the properties of azulenes. The configuration of π-electron system over the seven and five azulene rings develops a dipole moment that favors a displacement of the electrons towards an acceptor substituted in position 1 of the azulene. In this way the seven-ring adopts the stable structure of tropylium ion and consequently stabilizes the entire system in which it is involved. This explains the ease of azulenes to react in electrophilic substitutions as well as to donate electrons to acceptor systems. This paper is focused on the donor property of azulene and is limited to the most relevant acceptors, namely methylium ions and positive charged heteroaryls. Several cases of neutral molecules benefiting from azulen-1-yl assistance are also quickly described. It was considered useful to briefly summarize the synthesis of the compounds considered. The change in the charge distribution on the studied systems results in variations in the chemical shifts of protons and carbon atoms in the NMR spectra as well as some changes in the UV-Vis spectra. The pKR+ value of some pseudobases, such as cations, is a valuable indicator of their stability. The analysis of the mentioned parameters highlights the role of azulene as an electron donor on the stability of the systems in which it is included especially in those with positive charge. Certainly this property of azulene will further stimulate the building of chemical systems belonging to a series of areas of theoretical interest but also in those of technical interest such as dyes, active materials for electronic, optoelectronic and electrochromic devices.
This entry is adapted from the peer-reviewed paper 10.3390/sym13040526
References
- Solà, M. Forty years of Clar’s aromatic π-sextet rule. Front. Chem. 2013, 1, 22.
- Dauben, H.J., Jr.; Bertelli, D.J. Heptalene. J. Am. Chem. Soc. 1961, 83, 4659–4660.
- Bally, T.; Chai, S.; Neuenschwander, M.; Zhu, Z. Pentalene: Formation, Electronic, and Vibrational Structure. J. Am. Chem. Soc. 1997, 119, 1869–1875.
- Mikheev, Y.A.; Guseva, L.N.; Ershov, Y.A. Nature of the aromaticity of azulene and the dimers responsible for its chromaticity. Russ. J. Phys. Chem. 2012, 86, 1875–1880.
- Tobe, Y. Non-Alternant Non-Benzenoid Aromatic Compounds: Past, Present, and Future. Chem. Rec. 2014, 15, 86–96.
- Fischer, L.J.; Dutton, A.S.; Winter, A.H. Anomalous effect of non-alternant hydrocarbons on carbocation and carbanion electronic configurations. Chem. Sci. 2017, 8, 4231–4241.
- Ito, S.; Morita, N.; Asao, T. Azulene analogues of triphenylmethyl cation; extremely stable hydrocarbon cations. Tetrahedron Lett. 1991, 32, 773–776.
- Franke, H.; Mȕhlstädt, M. Substitution am azulen. Z. Chem. 1962, 2, 275–276.
- Franke, H.; Mȕhlstädt, M. Substitution am azulen. I. J. Prakt. Chem. 1967, 35, 249–261.
- Franke, H.; Mȕhlstädt, M. Substitution am azulen. II. J. Prakt. Chem. 1967, 35, 262–270.
- Zeller, K.-P. Houben Weyl, Methoden der Organischen Chemie; G. Thieme Verlag: Stuttgart, Germany; New York, NY, USA, 1985; Volume V/2c, pp. 258–263.
- Kirby, E.C.; Reid, D.H. Conjugated Cyclic Hydrocarbons and Their Heterocyclic Analogues. The Condensation of Azulenes with Homocyclic and Heterocyclic Aromatic Aldehydes in the Presence of Perchloric Acid. Part II. J. Chem. Soc. 1960, 494–501.
- Kirby, E.C.; Reid, D.H. Conjugated Cyclic Hydrocarbons and Their Heterocyclic Analogues. The Condensation of Azulenes with aliphatic aldehydes in the presence of perchloric acid. Part VI. J. Chem. Soc. 1961, 3579–3593.
- Hafner, K.; Pelster, H.; Schneider, J. Zur kenntnis der azuleneVII. 1- bzw. 3-alkyliden-azulenium salze. Justus Liebigs Ann. Chem. 1961, 650, 62–80.
- Asao, T. Azulenic novel π-electronic compounds. Pure Appl. Chem. 1990, 62, 507–512.
- Ito, S.; Morita, N.; Asao, T. Tris(3,6-di-t-butyl-1-azulenyl)methyl Cation; Hydrocarbon with the highest pKR+. Tetrahedron Lett. 1994, 35, 751–754.
- Ito, S.; Morita, N.; Asao, T. Dynamic stereochemistry of tri(2-methyl-1-azulenyl)methyl cation; steric effect of 2-methyl groups on rotational barriers and mechanism. Tetrahedron Lett. 1994, 35, 3723–3726.
- Ito, S.; Fujita, M.; Morita, N.; Asao, T. Synthesis and Dynamic Stereochemistry of Bis(3-methyl-1-azulenyl)(1-naphthyl)methyl Hexafluorophosphate. Clear Evidence of the One-Ring Flip Mechanism of Molecular Propeller by Controlling the Flipping Ring. Chem. Lett. 1995, 475–476.
- Ito, S.; Morita, N.; Asao, T. Synthesis and Dynamic Stereochemistry of Tris(2-methyl-1-azulenyl)methyl Cation and the Corresponding Methane Derivative. Evidence That the Conjugative Effect Largely Contributes to the Transition State of the Ring Flipping. Bull. Chem. Soc. Jpn. 1995, 68, 2639–2648.
- Ito, S.; Fujita, M.; Morita, N.; Asao, T. Synthesis and Dynamic Stereochemistry of Di-1-azulenyl(1- and 2-naphthyl)methyl Hexafluorophosphates. Clear Evidence of the One-Ring Flip Mechanism of a Molecular Propeller by Controlling the Flipping Ring. Bull. Chem. Soc. Jpn. 1995, 68, 3611–3620.
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. Syntheses of Azulene Analogues of Triphenylmethyl Cation: Extremely Stable Hydrocarbon Carbocations and the First Example of a One-Ring Flip as the Threshold Rotation Mechanism for Molecular Propellers. Bull. Chem. Soc. Jpn. 1995, 68, 1409–1436.
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. The Most Stable Methylcation. Tris(6-methoxy-1-azulenyl)methyl Hexafluorophosphat. Chem. Lett. 1996, 175–176.
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. Synthesis and Properties of Extremely Stable Tris(6-methoxy-1-azulenyl)methyl Cation and a Series of Di(1-azulenyl)phenylmethyl and (1-Azulenyl)diphenylmethyl Cations Stabilized by Methoxy Substituents. Bull. Chem. Soc. Jpn. 1999, 72, 839–849.
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. Syntheses and Properties of Extremely Stable Di(1-azulenyl)phenylmethyl and (1-Azulenyl)diphenylmethyl Cations Having Dimethylamino Substituents on Their Phenyl Groups. Bull. Chem. Soc. Jpn. 1996, 69, 3225–3237.
- Ito, S.; Kikuchi, N.; Morita, N.; Asao, T. Tris[6-(dimethylamino)-1-azulenyl]methyl Hexafluorophosphate. Extremely Stable Methyl Cation with the Highest pKR+Value. J. Org. Chem. 1999, 64, 5815–5821.
- Deno, N.C.; Schriesheim, A. Carbonium Ions. II. Linear Free Energy Relationships in Arylcarbonium Ion Equilibria. J. Am. Chem. Soc. 1955, 77, 3051–3054.
- Ito, S.; Kikuchi, S.; Okujima, T. Cations and Dications Composed of Two Di(1-azulenyl)methylium Units Connected with 2,5-Thiophenediyl and 2,5-Thienothiophenediyl Spacers. J. Org. Chem. 2001, 66, 2470–2479.
- Kikuchi, S.; Iki, M.; Ikeda, C.; Imafuku, K. Synthesis and Properties of (1-Azulenyl)di(3-indolyl)methylium Hexafluorophosphates. Heterocycles 2005, 66, 275–283.
- Takekuma, S.; Sasaki, M.; Takekuma, H.; Yamamoto, H. Preparation and Characteristic Properties of 1,4-Bis(3-guaiazulenylmethylium)benzene Bishexafluorophosphate. Chem. Lett. 1999, 28, 999–1000.
- Ito, S.; Morita, N.; Asao, T. Synthesis, Properties, and Redox Behaviors of Di- and Trications Composed of Di(1-azulenyl)methylium Units Connected byp- andm-Phenylene and 1,3,5-Benzenetriyl Spacers. Bull. Chem. Soc. Jpn. 2000, 73, 1865–1874.
- Deuchert, K.; Hünig, S. Multistage Organic Redox Systems-A General Structural Principle. Angew. Chem. Int. Ed. Engl. 1978, 17, 875–886.
- Ito, S.; Morita, N.; Asao, T. Syntheses, Properties, and Redox Behaviors of Di(1-azulenyl)ferrocenylmethyl Cations and 1,3-Bis[(1-azulenyl)ferrocenylmethylium]azulene Dication. J. Org. Chem. 1996, 61, 5077–5082.
- Shoji, T.; Ito, S. The Preparation and Properties of Heteroarylazulenes and Hetero-Fused Azulenes. Adv. Heterocycl. Chem. 2018, 1–54.
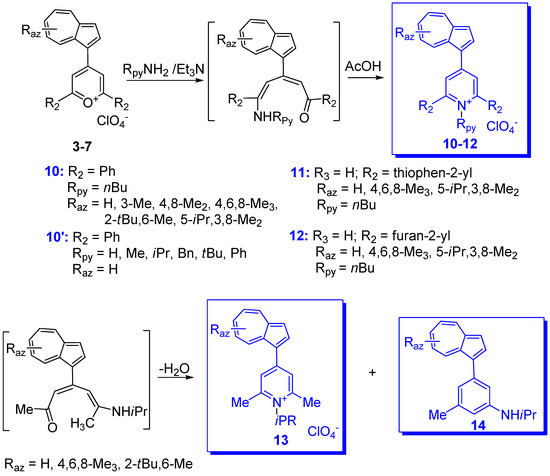
- Razus, A.C.; Birzan, L. Synthesis of azulenic compounds substituted in the 1-position with heterocycles. Monatsh. Chem. 2018, 150, 139–161.
- Krivun, S.V.; Baranov, S.N.; Buryiak, A.I. Introduction of the pyrylium ring into compounds of the aromatic and heterocyclic series. Chem. Heterocycl. Compd. 1971, 7, 1233–1236.
- Dorofeenko, G.N.; Koblik, A.V.; Polyakova, T.I.; Murad’yan, L.A. Synthesis of 6-hetaryl-substituted azulenes and their reactions with 2,6-diphenylpyrylium perchlorate. Chem. Heterocycl. Compd. 1980, 16, 807–810.
- Razus, A.C.; Birzan, L.; Pavel, C.; Lehadus, O.; Corbu, A.C.; Enache, C. Azulene-substituted Pyranylium Salts. Syntheses and Products Characterization. J. Heterocycl. Chem. 2006, 43, 963–977.
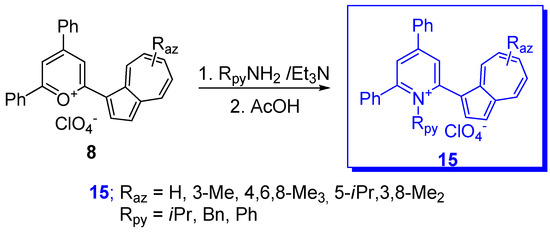
- Razus, A.C.; Birzan, L.; Zaharia, O.C. Enache. 2-(Azulen-1-yl)-4,6-diphenyl Substituted Six-membered Heteroaromatics. J. Heterocycl. Chem. 2008, 45, 1139–1147.
- Said, S.A.; Fiksdahl, A. Stereoselective transformation of amines via chiral 2,4,6-triphenylpyridinium intermediates. Tetrahedron Asymmetry 2001, 12, 1947–1951.
- Razus, A.C.; Birzan, L.; Pavel, C.; Lehadus, O.; Corbu, A.; Chiraleu, F.; Enache, C. Azulene-substituted Pyridines and Pyridinium Salts. 1. Azulene-substituted Pyridinium salts. J. Heterocycl. Chem. 2007, 44, 245–251.
- Razus, A.C.; Birzan, L.; Pavel, C.; Lehadus, O.; Corbu, A.; Chiraleu, F.; Enache, C. Azulene-substituted Pyridines and Pyridinium Salts. 2. J. Heterocycl. Chem. 2007, 44, 251–260.
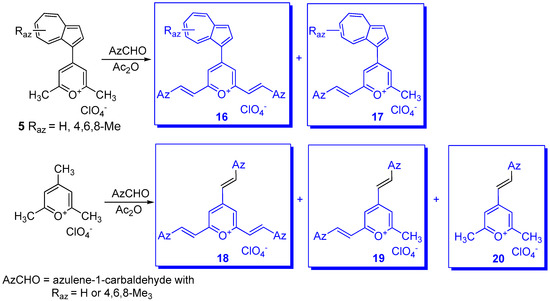
- Razus, A.C.; Birzan, L.; Cristea, M.; Tecuceanu, V.; Hanganu, A.; Enache, C. 4-(Azulen-1-yl) six-membered heteroaromatics substituted with thiophen-2-yl or furan-2-yl moieties in 2 and 6 positions. J. Heterocycl. Chem. 2011, 48, 1019–1027.
- Razus, A.C.; Cristian, L.; Nica, S.; Zaharia, O.; Hanganu, A. Pyrylium salts with 2-(azulene-1-yl) vinyl substituents in 2-, 4- and/or 6-positions. Arkivoc 2011, 9, 38–50.
- Birzan, L.; Cristea, M.; Draghici, C.; Tecuceanu, V.; Hanganu, A.; Ungureanu, E.-M.; Razus, A.C. 4-(Azulen-1-yl) six-membered heteroaromatics substituted in 2- and 6- positions with 2-(2-furyl)vinyl, 2-(2-thienyl)vinyl or 2-(3-thienyl)vinyl moieties. Tetrahedron 2017, 73, 2488–2500.
- Dykes, G.; Boruski, S.; Heiermann, J.; Korning, J.; Opwis, K.; Henkel, G.; Kocerling, M. First intermolecular palladium catalyzed arylation of an unfunctionalized aromatic hydrocarbon. J. Organomet. Chem. 2000, 606, 108–110.
- Rasala, D.; Bak, T.; Kolehmainen, E.; Styrcz, S.; Gawinecki, R. Transmission of electronic effects in 4-aryl-2, 6-diphenylpyrylium perchlorates and related compounds. J. Phys. Org. Chem. 1996, 9, 631–638.
- Detty, M.R.; McKelvey, J.M.; Luss, H.R. Tellurapyrylium dyes. The electron-donating properties of the chalcogen atoms to the chalcogenapyrylium nuclei and their radical dications, neutral radicals, and anions. Organometallics 1988, 7, 1131–1147.
- Takekuma, S.; Mizutani, K.; Inoue, K.; Nakamura, M.; Sasaki, M.; Minematsu, T.; Takekuma, H. Reactions of azulene and guaiazulene with all-trans-retinal and trans-cinnamaldehyde: Comparative studies on spectroscopic, chemical, and electrochemical properties of monocarbenium-ions stabilized by expanded π-electron systems with an azulenyl or 3-guaiazulenyl group. Tetrahedron 2007, 63, 3882–3893.
- Farrell, T.; Meyer-Friedrichsen, T.; Malessa, M.; Haase, D.; Saak, W.; Asselberghs, I.; Wostyn, K.; Clays, K.; Persoons, A.; Heck, J.; et al. Azulenylium and guaiazulenylium cations as novel accepting moieties in e. xtended sesquifulvalene type D-A NLO chromophores. J. Chem. Soc. Dalton Trans. 2001, 29–36.
- Kerber, R.C.; Hsu, C.-M. Substituent effects on cyclopropenium ions. J. Am. Chem. Soc. 1973, 95, 3239–3245.