Nuclear factor-erythroid 2-related factor 2 (NRF2) and its major negative modulator Kelch-like ECH-associated protein 1 (KEAP1) are main players of the cellular defense mechanisms against internal and external cell stressors.
- Nrf2
- Keap1
- cancer
- oxidative stress
1. Introduction
Cancer is a non-communicable disease with an increasing incidence and mortality in many countries worldwide, which is expected to become the leading cause of death in every continent by the end of this century. According to the global statistical data published by (World Health Organization (WHO), Geneva, Switzerland) the main reason behind the growth of cancer-related deaths is the expansion of the world’s aging population [1]. Another critical phenomenon is that common cancer profiles have been changing in a way that infection and or poverty-related cancers tend to decrease while cancers that are associated with Westernized lifestyle tend to augment [2]. In this context, it is of great importance that researchers define the roles of critical molecular players related to tumor formation, progression, and metastasis. At the molecular level, cell division and death of damaged/mutated cells are tightly controlled by several pathways in order to prevent survival of damaged cells bearing mutations and pass those mutations to the next generations. Sometimes a critically damaged cell can take a life-changing decision and takes steps to secure its own survival despite the cost of transforming into a tumor cell. The steps to be taken in the way of transformation by a precancerous cell are described in the highly cited Weinberg review in detail [3]. Genes and signal transduction pathways common to more than one hallmark of cancer recently gained extra attention as promising therapeutic targets. Among the others, redox signaling emerged as a signal transduction pathway involved in every step of carcinogenesis [4]. Nuclear factor erythroid 2-related factor 2 (NRF2), also known as NFE2L2, is considered as the leading transcription factor controlling cellular redox homeostasis and antioxidant pathways [5]. Being the major stress regulator of the cell, NRF2 is involved in tumor formation, progression, and metastasis [6]. For this reason, a large number of studies is currently ongoing to better characterize NRF2 pathway and its roles in cancer. NRF2 displays a complex behavior in carcinogenesis [7]. To fully address the importance of NRF2 pathway in cancer, it is necessary to provide a description of its negative regulator KEAP1, that interacts with NRF2 to downmodulate its expression in cells and strictly control cellular homeostasis [8]. Under normal or low/moderate stress conditions, there is a tight balance between KEAP1 activity and NRF2 protein levels, which provides regulated antioxidant response, detoxification, and prevention of cancer. However, excessive stress, continuous overexpression of NRF2 or downregulation of KEAP1 cause a shift in this balance, which, in turn, acts in favor of carcinogenesis. Unraveling these roles would provide researchers to target NRF2 pathway in a more selective way to fully eradicate cancer without promoting its pro-oncogenic functions also known as the “dark side” of NRF2 [9]. In this respect, experimental studies wherein NRF2 function was abrogated with genetic or chemical approaches, support the notion that this transcription factor plays a cytoprotective role, acting as a tumor suppressor in specific contexts [10,11]. It has also been reported that NRF2 loss is strongly associated with tumor malignancy and metastatic behavior of cancer cells [12]. Moreover, partial or complete depletion of KEAP1 has been shown to promote cancer initiation and growth suggesting that KEAP1 can be also regarded as a tumor suppressor, similarly to NRF2 [13,14]. Based on collective data on NRF2 and KEAP1, there is a growing interest in better defining the therapeutic use of natural obtained or chemically synthesized activators of NRF2 with tumor-suppressing properties [15]. Yet, none of these compounds, either from natural or chemical sources, showed a consistent, stable, and dose-dependent effect to become a solid anticancer drug candidate, due to the context-dependent effects of NRF2 pathway in tumors [16]. Moreover, many studies reported that the abnormal activation of NRF2 is a common event in tumor cells, caused by several factors like somatic mutations, oncogenic signaling, epigenetic changes, metabolic reprograming and altered redox balance in cancer cells [17]. Indeed, abnormal NRF2 expression has been detected in various tumors such as lung, esophageal, laryngeal, skin, pancreas and liver cancers [18]. Despite these observations might argue against the assumption of NRF2 being a tumor suppressor, they actually indicate that NRF2, is a context-dependent transcription factor that can act as an oncogene under certain circumstances. In addition, abnormal KEAP1 expression has been observed in several cancers including lung, liver, pancreas, and ovarian cancers [19]. Thus, based on these data, it appears that there is a fine-tuning between KEAP1 and NRF2 levels and this determines which effect of this pathway will be more prominent under specific circumstances of a certain type of tumor. On the other hand, extensive research is focusing on NRF2 inhibitors in consideration of its cancer promoting roles, especially in the later stages of tumorigenesis [16]. However, these inhibitors may lack specificity and therefore interact with other downstream pathways causing undesired effects. Thus, it is of great importance that these drug candidates will be meticulously tested in large clinical studies in order to identify the specific cohorts of patients and the clinical context most likely having beneficial effects and minimal side toxicity.
2. NRF2 and KEAP1 Signaling Pathway
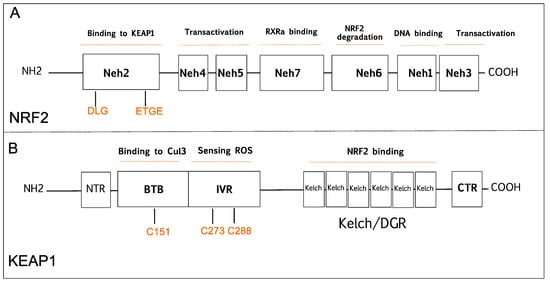
2.1. The Tumor Suppressive Role of NRF2 Pathway
2.2. The Tumor Suppressive Role of KEAP1
2.3. The Carcinogenic Role of NRF2
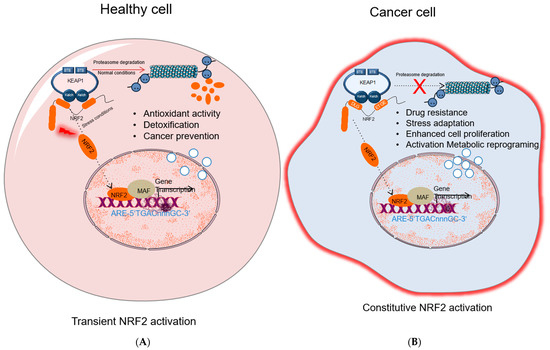
2.4. The Carcinogenic Role of KEAP1
3. NRF2 Activation Mechanisms in Cancer
Comprehensive studies validated that NRF2-KEAP1 signaling pathway is activated in several cancers such as skin, lung, bladder, hepatocellular carcinoma, esophagus, ovarian, prostate, pancreatic, and breast cancer [6,52,53,55,57]. The molecular mechanisms responsible for the activation of NRF2 in cancer are schematized in Figure 3 and further details are discussed below:
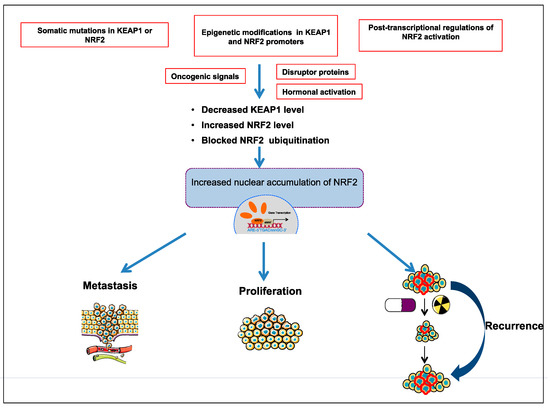
3.1. The Somatic Mutations in KEAP1 or NRF2
3.2. Epigenetic Modifications in KEAP1 and NRF2 Promoters
3.3. Post-Transcriptional Regulation of NRF2 Activation
3.4. Disruptor Proteins
3.5. Oncogenic Signals
3.6. Hormonal Activation
Several studies validated the effects of hormonal activation of NRF2 on cancer progression. Gonadotropins and sex steroid hormones, including follicle-stimulating hormone (FSH), estrogen (E2), and luteinizing hormone (LH), have been reported to be critical in activation of NRF2 through the induction of ROS that inhibit KEAP1 by oxidation of its cysteine residues [88]. In addition, follicle-stimulating hormone (FSH) is known to induce expression of vascular endothelial growth factor (VEGF) and hypoxia inducible factor 1α (HIF1α). Thus, FSH contributes to tumor angiogenesis through ROS-mediated NRF2 signaling [89].
This entry is adapted from the peer-reviewed paper 10.3390/molecules26051417