Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
The magnitude of the host immune response can be regulated by either stimulatory or inhibitory immune checkpoint molecules. Receptor-ligand binding between inhibitory molecules is often exploited by tumours to suppress anti-tumour immune responses. Immune checkpoint inhibitors that block these inhibitory interactions can relieve T-cells from negative regulation, and have yielded remarkable activity in the clinic.
- hypoxia
- immune suppression
- hypoxia-activated prodrug
- tarloxotinib
1. Introduction
Immune checkpoint inhibitors (ICIs) sit at the forefront of cancer immunotherapy. They are emerging as the primary treatment option for many advanced stage cancer patients as a result of their clinical success. Within the last decade, seven ICIs targeting the immune checkpoint receptors cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed death protein 1 (PD-1), and the ligand to the checkpoint receptor PD-1 programme death protein ligand 1 (PD-L1) have been approved by the FDA for the treatment of a variety of malignancies [1]. While this remarkable progress has revolutionised the field of immuno-oncology, in reality, only a subset of cancer types currently respond to ICI treatments, with patients that gain long-term benefits remaining in the minority, reflected by objective response rates between 10 and 40% [2][3][4][5][6].
In general, immunotherapies are designed to increase the number and functionality of anti-tumour effector cells. The majority of such treatments target activating antigen-specific T-cells to selectively eliminate tumours through the recognition of unique or aberrantly expressed antigens presented as peptides by major histocompatibility complex (MHC) molecules on the tumour cell surface. ICIs such as anti-CTLA-4, anti-PD-1 and anti-PD-L1 monoclonal antibodies relieve T-cells from negative regulation governed by immune checkpoints by blocking the interactions between immune checkpoint receptors and their respective ligands on tumour cells, infiltrating myeloid cells or T-cells themselves [7]. The immune cell composition within the tumour microenvironment varies between patients (and sometimes between individual tumour lesions). Some have a “cold” immune privileged tumour phenotype, with minimal infiltration of immune cells (e.g., reduced numbers of effector T cells, natural killer cells and antigen presenting cells) that do not respond well to ICIs, while others possess a “hot” immune-infiltrated tumour phenotype that can respond well to ICIs [8][9]. This phenomena is suggestive of underlying resistance mechanisms that limit further advances of ICIs as monotherapy [10][11].
Tumours with high expression of PD-L1 respond better to ICIs targeting the PD-1/PD-L1 pathway and are also found to carry more tumour somatic mutations [12][13]. This suggests that the extent of mutagenesis in cancer cells correlates with the degree of immunogenicity of a given tumour type. A higher frequency of mutations in cellular DNA creates a greater chance of these cells at generating neoantigens which will be recognised as foreign and targeted by antigen presenting cells (APCs). Patients bearing tumours with a high mutational load are more responsive to ICIs due to increased tumour immunogenicity. For example, neoantigen signature in tumours correlated well with overall survival in patients undergoing anti-CTLA-4 treatment [12][14]. However, any increased immunogenicity can be offset by upregulating tumour expression of PD-L1 to evade immune destruction. Therefore, tumours often cannot be eradicated by the immune response.
Tumour resistance to ICIs can be classified into three broad categories: (i) primary resistance, in which an anti-tumour immune response cannot be elicited in the patient; (ii) adaptive immune resistance, in which an active anti-tumour immune response is present and can recognise cancerous cells but is unable to eliminate them due to immune evasion mechanisms established within the tumour milieu; and (iii) acquired resistance, in which the tumour initially responds to immunotherapy but subsequently becomes resistant and progresses on therapy [10][11][15]. There are numerous intrinsic and extrinsic factors that contribute to resistance to ICI treatment in patients, such as mutational load within the tumour and thus availability of tumour-associated antigens, central and peripheral tolerance mechanisms that limit the T-cell repertoire to tumour antigens, immunologically ignored tumour antigens, tumour microenvironment-associated factors (e.g., level and character of immune infiltrate, and immunosuppressive features such as tumour hypoxia), environmental factors (e.g., diet and microbiota that can alter capacity for immunity), endocrine and metabolic factors (e.g., stress response, obesity and individual variability in pharmacokinetics of treatment agents), and demographical factors (e.g., impact of sex and age on immunity) [10][11][16][17]. It is necessary for the immune response to overcome these immunosuppressive mechanisms mediated by the tumour and its microenvironment to elicit robust and durable anti-cancer immunity.
2. Tumour Hypoxia and Hypoxia-Mediated Immunosuppression
2.1. Tumour Hypoxia
Tumour hypoxia, defined as a state in which the oxygen levels are less than 1% O2 (10,000 ppm O2; 10 μM O2), is a pervasive feature of human tumours. It arises due to the abnormal structure of the tumour vasculature, leading to a mismatch between oxygen delivery and consumption [18][19]. Unlike normal tissues, the tumour vasculature lacks an organised network and is hyper-permeable, dilated and convoluted with areas of occlusions leading to poor or fluctuating blood perfusion. The rapid rate of cellular proliferation results in tumour cells also arising at an increased distance from functional blood vessels, with tumours larger than 2 mm experiencing limited oxygen and nutrient supply as well as acidosis due to inadequate waste exchange [20][21]. Cells located near the diffusion limit of oxygen (approximately 200 µm away from the blood vessels) are therefore quiescent (not rapidly proliferating) and experience diffusion-limited chronic hypoxia (Figure 1). In contrast, transient perfusion-limited hypoxia or intermittent hypoxia is caused by temporary blockage of the tumour vasculature [22]. Hypoxic tumours upregulate their expression of vascular endothelial growth factor (VEGF) to promote angiogenesis to allow for the formation of new blood vessels from existing ones to promote tumour growth and adapt to the microenvironment with limited supply of oxygen and nutrients [18][23].
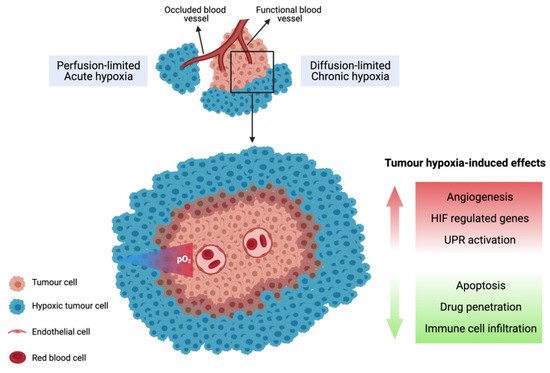
Figure 1. Illustration of chronic and acute tumour hypoxia. Diffusion-limited hypoxia occurs when cells are located near the diffusion limit of oxygen. Perfusion-limited hypoxia arises due to temporary occlusion of tumour vasculature.
2.2. Biological Response to Tumour Hypoxia
The hypoxic response is primarily governed by hypoxia-inducible factor (HIF)-1 transcription factor, which is a heterodimer consisting of a labile HIF-1α subunit and a constitutive HIF-1β subunit. The HIF-1α subunit is unstable under oxygenated conditions due to its regulation by prolyl hydroxylase-domain protein 1-3 (PHD1-3), which hydroxylates the oxygen-dependent degradation domains (ODD) of HIF-1α using molecular oxygen as a substrate. The hydroxylated prolines 402 and 564 are recognised by the von Hippel-Lindau (VHL) complex to target HIF-1α for degradation by the proteasome. Under hypoxic conditions, the ODD cannot be hydroxylated by PHDs, which leads to accumulation of HIF-1α and the formation of the active transcription factor HIF-1. The active HIF-1 heterodimer binds to hypoxia-responsive elements in the promoter/enhancer regions of HIF-1 regulated genes to transcriptionally activate a wide variety of genes involved in cell metabolism, angiogenesis, glucose metabolism, apoptosis, cell survival, cell proliferation and pH regulation [24][25][26]. Another HIF family transcription factor, HIF-2α, is also involved in activating hypoxic responses and is structurally similar to HIF-1α apart from the transactivation domain. This difference confers target gene specificity of HIF-1α and HIF-2α, leading to them having both overlapping and unique target genes [27][28][29]. Unlike HIF-1α, which is the main regulator of the glycolytic pathway and expressed mainly during the acute phase of hypoxia response (<24 h), HIF-2α is mostly involved in promoting an undifferentiated phenotype of pluripotent cells and drives the chronic response of hypoxia (>24 h) [30][31].
The unfolded-protein response (UPR) is also an important adaptation to severe hypoxia (reviewed in [32][33][34]). Prolonged exposure of cells to hypoxia results in endoplasmic reticulum (ER) stress and disruptions in protein folding and trafficking as the cells attempt to survive under hypoxia [32]. The accumulation of unfolded or misfolded proteins in the ER induces higher demand of binding immunoglobin protein (BiP), which initiates the UPR by dissociating from luminal domains of proteins including protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6) [33]. Upon dissociation, PERK and IRE1α become activated via multimerization and autophosphorylation. Activated PERK phosphorylates the eukaryotic initiation factor eIF2, leading to the translation of activating transcription factor 4 (ATF4). ATF4 then induces genes involved in oxidative stress resistance, redox homeostasis and amino acid biosynthesis [33][35]. Activated IRE1α degrades mRNAs and performs splicing of X-box binding protein transcription factor (XBP1) mRNA to generate the transcriptionally active spliced XBP1 (XBP1s). Both ATF4 and XBP1s then induce transcriptional programs to restore ER homeostasis [36]. ATF6, on the other hand, is translocated to the Golgi apparatus where it is processed by proteases to release a cytoplasmic domain ATF6f (p50). The transcriptionally active ATF6f is released to then activate a transcriptional program to restore ER homeostasis and survival [33][37]. Hypoxia-induced UPR can support the survival of cancer cells, promote angiogenesis and promote cancer cell resistance to chemotherapy [32][38].
Many tumour types contain high fractions of hypoxia such as those of the brain, head and neck, lung, breast, prostate, pancreas and cervix [18][19][39]. The relationship between tumour hypoxia and poor prognosis is firmly established, being associated with resistance to radiotherapy, chemotherapy and immunotherapy [16][18][40][41][42][43][44]. Hypoxia can affect many aspects of the tumour biology, such as the promotion of invasiveness and metastasis [45][46], suppression of apoptosis [47], induction of tumour angiogenesis [48] and altered tumour cell metabolism [49]. Moreover, it has been recognised that hypoxia is central to the generation of an immunosuppressive TME, thereby inhibiting the anti-tumour immune response [16][28][50]. As detailed in this review, tumour hypoxia can drive the suppression of the function and proliferation of effector T-cells and exacerbate tumour escape from immune surveillance, with a network of immunosuppressive cells, growth factors and cytokines being implicated in this process [16].
2.3. Hypoxia Promotes Immune Tolerance through Multiple Mechanisms
Tumours can evade immune recognition and destruction by cytotoxic T-cells via numerous mechanisms, including the generation of an immunosuppressive environment and development of resistance to clearance by immune effector cells [51]. An immunosuppressive environment manifests through the recruitment of immunosuppressive cells such as regulatory T-cells (Tregs) and myeloid derived suppressor cells (MDSCs), which suppress the effector function of cytotoxic T-cells through the production of suppressive factors, including tumour growth factor (TGF)-β, IL-10, VEGF, indoleamine 2, 3-dioxygenase (IDO) and arginase. These proteins, growth factors and cytokines can also promote tumour invasiveness, angiogenesis, and proliferation, as well as preventing the full activation and maturation of APCs. Immature DCs, in the absence of co-stimulatory molecules such as CD80, CD86 and CD40, promote T-cell tolerance rather than activation [51][52][53][54][55]. Similarly, tumour cells that lack the expression of co-stimulatory molecules can also induce T-cell tolerance or anergy when T-cells engage with tumour antigens present on the cell surface [56]. Further, the recruited MDSCs along with tumour associated macrophages (TAMs) create an inflammatory TME which facilitates tumour formation, progression, angiogenesis and metastasis by inducing chronic inflammation at tumour sites [51][57]. Tumours also develop resistance to cytotoxic T-cell killing by creating a defective antigen presentation pathway through the down-regulation of MHC molecules, transporter associated with antigen processing protein (TAP), and the tumour antigen itself. Defects in the antigen presentation machinery consequently lead to impaired tumour clearance and enhanced tumour progression, as cytotoxic T-cells can no longer recognise tumour antigens and exert their cytotoxic functions. Such tumour cells lack an immunogenic epitope and are thus ignored by the immune system, leading to a selective survival advantage [51][58][59][60]. The immune system itself can also contribute to tumour immune evasion through “immunoediting”, in which the immune system selectively eliminates immunogenic tumour cells, resulting in the survival of immune-resistant cancer cell clones [61][62].
Accumulating evidence indicates that most of these immunosuppressive mechanisms are orchestrated by tumour hypoxia through a network of immunosuppressive soluble factors and regulatory cell populations [16][18][19]. Specifically, tumour hypoxia has been shown to attract immunosuppressive Tregs [63], regulate the maturation and function of MDSCs [64], entrap and re-educate macrophages toward an immunosuppressive M2-like phenotype [65], and severely reduce the function of activated T-cells as a consequence of adenosine accumulation (Figure 2) [66]. Crosstalk between recruited regulatory cell populations also amplifies the production of immunosuppressive cytokines such as IL-10 and TGF-β [67]. Significantly, these effects can be reversed directly by hyperoxic breathing in animal models, including the restoration of activated T-cell infiltration and heightened release of pro-inflammatory cytokines and chemokines [68], indicating therapeutic interventions that suppress tumour hypoxia may hold considerable promise.
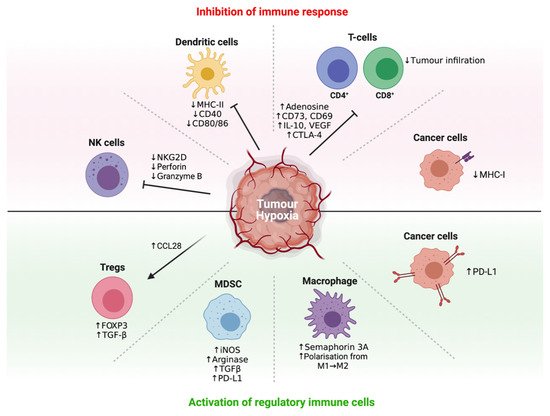
Figure 2. Regulation of immune response by tumour hypoxia. Different cell types by which tumour hypoxia influences in the tumour microenvironment.
2.4. Hypoxia Recruits Immunosuppressive Cells to Promote Immune Tolerance
Tregs are derived from naïve CD4+ T-cells under the influence of TGF-β or IL-2 cytokines. Tregs are characterised by the expression of the forkhead box P3 (FOXP3) transcription factor and cell-surface molecules such as CD25, CTLA-4 and LAG-3 [61][69]. The hypoxia-induced stabilisation of HIF-1 has been shown to upregulate FOXP3, which promotes the formation of Tregs from CD4+ T-cells [70]. Typically, Tregs impede T-cell responses and inflammation through the secretion of immunosuppressive cytokines such as IL-10 (inhibits expression of MHC molecules and co-stimulatory molecules on APCs) and TGF-β (inhibits T-cell proliferation), or via interaction between CTLA-4 on Tregs and CD80/86 on APCs (limits T-cell priming) or by sequestering IL-2 from naïve T-cells by virtue of the high affinity receptor CD25 on Tregs. Although Tregs are essential at maintaining self-tolerance to prevent autoimmunity, their accumulation in the tumour suppresses the anti-tumour immune response [61][69]. Hypoxia also promotes the recruitment of Tregs through HIF-1-mediated induction of the C-C motif chemokine ligand (CCL)-28. CCL28 acts as a chemoattractant for Tregs through its binding to C-C motif chemokine receptor 10 (CCR10) on Tregs. The expression of CCL28 has been found to correlate with HIF-1α expression in ovarian cancer and is associated with poor patient prognosis. Similarly, hypoxia-induced recruitment of Tregs has been associated with a poor prognosis for patients with hepatocellular carcinoma (HCC) and basal-like breast cancer [63][71][72]. HIF-2α is also involved in Treg stability as HIF-2α-knockout Tregs are functionally defective at suppressing effector T-cell function. Mice with FOXP3 conditional knockout of HIF-2α also showed resistance to the growth of MC38 colon tumours and metastatic invasion of B16.F10 melanoma [73].
Hypoxia regulates the function and maturation of MDSCs, a heterogeneous group of immature immune cells of myeloid origin, consisting of immature macrophages, granulocytes and DCs. MDSCs are generated from the bone marrow and have the ability to differentiate into mature myeloid cells in the presence of the appropriate cytokines. However, their maturation is restrained under hypoxia, resulting in the accumulation of immature MDSCs in the lymphoid tissues and the tumour, leading to suppression of appropriate immune responses [50][64]. This is due to HIF-1-induced upregulation of ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2), which converts extracellular adenosine triphosphate (ATP) to 5′-adenosine monophosphate (AMP). 5′-AMP prevents the maturation of MDSCs and promotes their maintenance [74]. Tumour-associated MDSCs also upregulate the production of nitric oxide (NO) and arginase-1, leading to antigen-specific Treg proliferation as well as the suppression of antigen-specific and non-specific T-cell functions. Hypoxia/HIF-1 induces the expression of tumour-derived factors such as VEGF, GM-CSF and prostaglandins, which further contribute to the accumulation of MDSCs in the TME. HIF-1-induced upregulation of CCL26 also increases the recruitment of MDSCs that express the cognate receptor, C-X3-C motif receptor 1 (CX3CR1) [50][64][75]. HIF-1 directly regulates the expression of PD-L1, and under hypoxic conditions, PD-L1 is upregulated on MDSCs to promote T-cell anergy and tolerance [50][76]. HIF-1 also promotes the differentiation of MDSCs into immunosuppressive TAMs that further dampen down the anti-tumour immune response [64][76].
Macrophages are derived from myeloid progenitor cells via a monocyte precursor. Following their infiltration into solid tumours, tumour-derived cytokines such as IL-4 and IL-10 can polarise these macrophages into a so-called M2-like phenotype (F4/80+ CD206+ CD11c-), giving rise to TAMs that are immunosuppressive (compared to its immunostimulatory M1-like phenotype counterpart (F4/80+ CD206- CD11c+)). M1-like macrophages are generally activated by IFN-γ and lipopolysaccharide (LPS), and produce high levels of IL-12 to promote the anti-tumour immune response. The abundance of M2-like macrophages in the hypoxic TME facilitates tumour progression through the production of high levels of IL-10, and by promoting angiogenesis, invasion and metastasis [77][78][79][80]. This maladaptive polarization of macrophages is intrinsically connected with the hypoxia/HIF sensors and the UPR [81]. Tumour hypoxia recruits M2-like macrophages via the HIF-1-regulated secretion of chemoattractant VEGF and endothelins, leading to their enhanced migration into the less vascularised regions of the tumour. Hypoxia-induced tumour-secreted Semaphorin 3A also contributes to M2-like macrophage recruitment to the hypoxic TME by binding to Neuropilin-1 expressed on macrophages [82]. HIF-2α is also involved in TAM accumulation in the TME and is stabilized in hypoxic macrophages. TAMs with high levels of HIF-2α correlate with increased tumour grade, and a high number of HIF-2α-expressing TAMs is associated with poor prognosis and tumour recurrence [83][84]. Further, in both murine HCC and colitis-associated cancer models, mice with HIF-2α-deficient TAMs showed reduced tumour infiltration of TAMs [85]. TAMs resident in the hypoxic areas of the tumour upregulate their expression of matrix metalloproteinase-7 protein, which cleaves Fas ligand from neighbouring cells rendering tumours less responsive to lysis by T-cells and natural killer (NK) cells [50][86]. TAMs express inducible nitric oxide synthase (iNOS) which produces NO and arginase-I, both of which suppress T-cell signal transduction and T-cell function and deplete the supply of L-arginine important for T-cell proliferation and survival. The production of iNOS and arginase-I is increased under hypoxia as their expression is mediated by HIF-1 at the transcriptional level, resulting in enhanced suppression of the anti-tumour immune response [87][88]. Not surprisingly, elevated numbers of TAMs are often associated with poor prognosis [82].
2.5. Hypoxia Interferes with and Suppresses Effector T-Cell, DC and NK Cell Function
Effector T-cell activity is disfavoured within the hypoxic tumour regions since HIF-1 acts as a negative regulator of effector T-cell activation and function. For example, hypoxia has been shown to reduce the expression of T-cell activation markers CD69 and CD40L [89] and studies have implicated HIF-1 in this process, as gene knockout οf HIF-1 in T-cells is sufficient to restore their proliferative phenotype and secretion of pro-inflammatory cytokines, e.g., IFN-γ [82][90]. In vitro assays have shown that T-cells cultured under 1–5% oxygen had a significant reduction in T-cell proliferative activity compared to T-cells cultured in more oxygenated conditions (21% oxygen). T-cells cultured in a lower oxygen environment also exhibited decreased IL-2 and IFN-γ production [89]. However, the precise oxygen concentration dependence of these effects is not well defined. The mechanisms by which HIF-1 suppresses the function of effector T-cells are complex, but include the upregulation of co-inhibitory receptors (e.g., CTLA-4 and LAG-3) [82][91], the differentiation of CD4+ T-cells into Tregs and the indirect effect of altered tumour cell metabolism [63][70]. As discussed earlier, hypoxia/HIF-1 transforms CD4+ T-cells into Tregs in a TGF-β-dependent manner [63][70]. The recruitment of Tregs into tumour sites suppresses the effector function of CD8+ T-cells. While a high effector T-cell/Treg ratio is favourable for the initiation of anti-tumour immune responses, the limited infiltration of CD4+ and CD8+ T-cells in hypoxic areas of the tumour leads to localised reductions in effector T-cell/Treg ratios and thus regional immunosuppression [16][43]. HIF-2α can also suppress T-cell function by upregulating PD-L1 expression on tumour cells. In patients with clear cell renal carcinoma, the expression of PD-L1 showed positive correlation with HIF-2α expression [92].
Changes in tumour cell metabolism also affect the function of effector T-cells within the hypoxic TME. Tumour cells often adapt to a hypoxic microenvironment by switching from oxidative phosphorylation to glycolysis, through HIF-1-mediated induction of various glycolytic enzymes to further elevate this process for ATP generation [93]. The elevated glycolytic activity in solid tumours leads to increased competition for nutrients between tumour and immune cells, as well as the increased production of glycolytic metabolites such as lactate, protons and carbonic acid, which promotes acidosis of the hypoxic TME [94][95][96]. The accumulation of lactic acid suppresses the proliferation and cytokine production activities of cytotoxic T-cells as well as inhibiting their cytolytic activity [97][98]. Further, the acidic TME impairs the secretion of proinflammatory cytokines by T-cells (e.g., IL-2, TNFs and IFN-γ) and upregulates CTLA-4 expression, rendering tumour infiltrating T-cells more susceptible to negative regulatory signals [99]. Thus, hypoxia-driven tumour acidosis promotes tumour progression and is a barrier to T-cell function in the TME.
The accumulation of extracellular adenosine is an important mechanism by which hypoxia can suppress T-cell activity. Dead and dying cells in the TME release ATP, which can be metabolised by ectonucleotidases CD73 and CD39 on the surface of immune cells. Critically, both ectonucleotidases are HIF-1 regulated, and their expression and activity is upregulated in hypoxic tumours, which leads to the increased production of cyclic adenosine monophosphate (cAMP) and adenosine, thereby enhancing immune suppression [82][100][101]. ATPs are first recognised and converted into AMPs by CD39, which are then converted by surface CD73 molecules into adenosines [82]. The cellular uptake of nucleosides is mediated by the human equilibrative nucleoside transporter 1 (ENT1), whose expression is reduced under hypoxic conditions, resulting in the accumulation of extracellular adenosine [102]. Adenosines can then bind to their receptors (A2AR and A2BR) on the immune cells to promote the production of intracellular cAMP, a factor that negatively regulates effector T-cell function and proliferation via diverse mechanisms. For example, cAMP can interfere with T-cell trafficking through the desensitisation of chemokine receptors and impairing the secretion of pro-inflammatory cytokines [43][103].
Effector T-cell infiltration into the tumour is hindered through the upregulation of VEGF to promote dysregulated angiogenesis, and via the downregulation of integrins (αLβ2) on vascular endothelium by upregulating IL-10 production [29][104]. Consequently, the abnormal, disorganised tumour neovasculature lacks the appropriate proteins for adhesion, attraction and extravasation of T-cells, leading to dysregulated trafficking of T-cells into the tumour bed. Furthermore, the enrichment of IL-10, VEGF and prostaglandin E2 under hypoxic conditions induces Fas-ligand expression on the tumour vasculature to promote T-cells apoptosis, ultimately leading to reduced T-cell accumulation within the TME [16]. Hypoxia via HIF-1 also reshapes the extracellular matrix by increasing collagen deposition and inducing stromal fibrosis, which also impede the accessibility of T-cells [29].
DC functions are also negatively influenced by hypoxia. Here, the expression of maturation and co-stimulatory molecules on DCs (e.g., CD40, CD80 and CD86) is downregulated, which negatively influences the activation of naïve T-cells [105]. The maturation and function of DCs are further affected by the hypoxia-induced upregulation of VEGF and IL-10 in the TME. VEGF inhibits the maturation of DCs, while IL-10 prevents the differentiation of monocytes into DCs and downregulates CCR7 expression, which alters the homing of DCs to the lymph nodes. Concurrently, hypoxia can upregulate the expression of PD-L1 on DCs to suppress T-cell function [16][50][82].
NK cell functions are also affected under hypoxic conditions. NK cells are cytotoxic lymphocytes belonging to the innate immune system that can directly lyse target tumour cells via the secretion of perforin and granzymes or via Fas/Fas-ligand-induced apoptosis [106]. Hypoxia/HIF can upregulate the expression of metalloproteinase ADAM10, which is responsible for the shedding of the ligand MHC-I polypeptide-related sequence A (MICA) from the surface of tumour cells. Surface MICA is a ligand for the activating receptor natural killer group 2 member D (NKG2D) on NK cells; however, soluble MICA can downregulate the expression of NKG2D on NK cells, which contributes to tumour immune evasion [107]. The expression of other activating receptors such as NKp30, NKp44 and NKp46 involved in target-recognition and killing is also down-regulated under hypoxic conditions [108]. Acidosis also increases the number of MDSCs in the tumour to inhibit the cytotoxicity of NK cells and facilitates tumour cell invasion, and is associated with poor patient prognosis [109][110]. Finally, hypoxia appears to also impair NKT cell activity through the HIF-2α-induced downregulation of Fas ligand expression and the upregulation of A2A receptors [111].
Collectively, preclinical data overwhelmingly indicate that tumour hypoxia plays a major role in regulating the function of immune cells and promoting an immunosuppressive tumour microenvironment. Clinical studies also support the association between hypoxia and immunosuppression. For example, in a cohort of 938 HNSCC patients, tumours enriched for hypoxia-responsive genes such as HIF1A, VEGF and carbonic anhydrase IX (CAIX) genes were strongly associated with the lack of CD8+ T-cell infiltrate and immune related gene signatures [112]. In a series of breast cancer surgical specimens, HIF-1 activity predicted the expression of immunosuppressive molecules including VEGF-A, IL-10 and TGF-β, and correlated with Treg infiltration [113]. The expression of HIF-1 was also positively correlated with Tregs and TGF-β has been observed in gastric cancer patient samples [114]. Further, the HIF-1-induced upregulation of VEGF has been found to directly impede T-cell activation in the ascites of ovarian cancer patients [115], leading to immune tolerance [116]. The upregulation of several gene expression clusters associated with tumour hypoxia was found in biopsy specimens of melanoma patients resistant to anti-PD-1 treatment [117]. It is notable that castration-resistant prostate cancer, colorectal cancer, and pancreatic cancer, all of which are frequently observed to be hypoxic, are typically resistant to ICI treatments [16].
This entry is adapted from the peer-reviewed paper 10.3390/cells10051006
References
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738.
- Yan, Y.; Kumar, A.B.; Finnes, H.; Markovic, S.N.; Park, S.; Dronca, R.S.; Dong, H. Combining Immune Checkpoint Inhibitors With Conventional Cancer Therapy. Front. Immunol. 2018, 9.
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76.
- Foley, K.; Kim, V.; Jaffee, E.; Zheng, L. Current progress in immunotherapy for pancreatic cancer. Cancer Lett. 2016, 381, 244–251.
- Patel, S.P. Immune checkpoint blockade for lung cancer: State of the art. Transl. Cancer Res. 2015, 4, 415–422.
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532.
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264.
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327.
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362.
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35.
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269.
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222.
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular Pathways: Next-Generation Immunotherapy—Inhibiting Programmed Death-Ligand 1 and Programmed Death-1. Clin. Cancer Res. 2012, 18, 6580.
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199.
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723.
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445.
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330.
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal. 2007, 9, 1221–1235.
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676.
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121.
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186.
- Horsman, M.R.; Mortensen, L.S.; Petersen, J.B.; Busk, M.; Overgaard, J. Imaging hypoxia to improve radiotherapy outcome. Nat. Rev. Clin. Oncol. 2012, 9, 674–687.
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713.
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472.
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014.
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732.
- Hu, C.J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol. Biol. Cell 2007, 18, 4528–4542.
- Wang, B.; Zhao, Q.; Zhang, Y.; Liu, Z.; Zheng, Z.; Liu, S.; Meng, L.; Xin, Y.; Jiang, X. Targeting hypoxia in the tumor microenvironment: A potential strategy to improve cancer immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 24.
- Pietrobon, V.; Marincola, F.M. Hypoxia and the phenomenon of immune exclusion. J. Transl. Med. 2021, 19, 9.
- Ratcliffe, P.J. HIF-1 and HIF-2: Working alone or together in hypoxia? J. Transl. Med. 2007, 117, 862–865.
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372.
- Bartoszewska, S.; Collawn, J.F. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell. Mol. Biol. Lett. 2020, 25, 18.
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102.
- Wouters, B.G.; Koritzinsky, M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer 2008, 8, 851–864.
- Rutkowski, D.T.; Kaufman, R.J. All roads lead to ATF4. Dev. Cell 2003, 4, 442–444.
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891.
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. ATF6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell Biol. 2000, 20, 5096–5106.
- Rzymski, T.; Milani, M.; Singleton, D.C.; Harris, A.L. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 2009, 8, 3838–3847.
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318.
- Hill, R.P.; Bristow, R.G.; Fyles, A.; Koritzinsky, M.; Milosevic, M.; Wouters, B.G. Hypoxia and Predicting Radiation Response. Semin. Radiat. Oncol. 2015, 25, 260–272.
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975.
- Dhani, N.; Fyles, A.; Hedley, D.; Milosevic, M. The Clinical Significance of Hypoxia in Human Cancers. Semin. Nucl. Med. 2015, 45, 110–121.
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230.
- Vuillefroy de Silly, R.; Dietrich, P.Y.; Walker, P.R. Hypoxia and antitumor CD8(+) T cells: An incompatible alliance? Oncoimmunology 2016, 5, e1232236.
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361.
- Lunt, S.J.; Chaudary, N.; Hill, R.P. The tumor microenvironment and metastatic disease. Clin. Exp. Metastasis 2009, 26, 19–34.
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol. Cell Biol. 2004, 24, 2875–2889.
- Harris, A.L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47.
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95.
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579.
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227.
- Munn, D.H.; Sharma, M.D.; Lee, J.R.; Jhaver, K.G.; Johnson, T.S.; Keskin, D.B.; Marshall, B.; Chandler, P.; Antonia, S.J.; Burgess, R.; et al. Potential Regulatory Function of Human Dendritic Cells Expressing Indoleamine 2,3-Dioxygenase. Science 2002, 297, 1867.
- Boutard, V.; Havouis, R.; Fouqueray, B.; Philippe, C.; Moulinoux, J.P.; Baud, L. Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. J. Immunol. 1995, 155, 2077–2084.
- Staveley-O’Carroll, K.; Sotomayor, E.; Montgomery, J.; Borrello, I.; Hwang, L.; Fein, S.; Pardoll, D.; Levitsky, H. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA 1998, 95, 1178–1183.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899.
- Maeurer, M.J.; Gollin, S.M.; Martin, D.; Swaney, W.; Bryant, J.; Castelli, C.; Robbins, P.; Parmiani, G.; Storkus, W.J.; Lotze, M.T. Tumor escape from immune recognition: Lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen. J. Clin. Investig. 1996, 98, 1633–1641.
- Garrido, F.; Ruiz-Cabello, F.; Cabrera, T.; Pérez-Villar, J.J.; López-Botet, M.; Duggan-Keen, M.; Stern, P.L. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol. Today 1997, 18, 89–95.
- Johnsen, A.K.; Templeton, D.J.; Sy, M.; Harding, C.V. Deficiency of transporter for antigen presentation (TAP) in tumor cells allows evasion of immune surveillance and increases tumorigenesis. J. Immunol. 1999, 163, 4224–4231.
- Murphy, K.; Weaver, C. Janeway’s Immunobiology, 9th ed.; Garland Science/Taylor & Francis Group, LLC: Brighton, UK, 2016.
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570.
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230.
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453.
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709.
- Adams, J.L.; Smothers, J.; Srinivasan, R.; Hoos, A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2015, 14, 603–622.
- Ostrand-Rosenberg, S.; Sinha, P.; Chornoguz, O.; Ecker, C. Regulating the suppressors: Apoptosis and inflammation govern the survival of tumor-induced myeloid-derived suppressor cells (MDSC). Cancer Immunol. Immunother. 2012, 61, 1319–1325.
- Hatfield, S.M.; Sitkovsky, M. Oxygenation to improve cancer vaccines, adoptive cell transfer and blockade of immunological negative regulators. Oncoimmunology 2015, 4, e1052934.
- Corthay, A. How do Regulatory T Cells Work? Scand. J. Immunol. 2009, 70, 326–336.
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793.
- Ren, L.; Yu, Y.; Wang, L.; Zhu, Z.; Lu, R.; Yao, Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget 2016, 7, 75763–75773.
- Yan, M.; Jene, N.; Byrne, D.; Millar, E.K.; O’Toole, S.A.; McNeil, C.M.; Bates, G.J.; Harris, A.L.; Banham, A.H.; Sutherland, R.L.; et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. BCR 2011, 13, R47.
- Hsu, T.-S.; Lin, Y.-L.; Wang, Y.-A.; Mo, S.-T.; Chi, P.-Y.; Lai, A.C.-Y.; Pan, H.-Y.; Chang, Y.-J.; Lai, M.-Z. HIF-2α is indispensable for regulatory T cell function. Nat. Commun. 2020, 11, 5005.
- Chiu, D.K.-C.; Tse, A.P.-W.; Xu, I.M.-J.; Di Cui, J.; Lai, R.K.-H.; Li, L.L.; Koh, H.-Y.; Tsang, F.H.-C.; Wei, L.L.; Wong, C.-M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517.
- Chiu, D.K.; Xu, I.M.; Lai, R.K.; Tse, A.P.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813.
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781.
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555.
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281.
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246.
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562.
- Díaz-Bulnes, P.; Saiz, M.L.; López-Larrea, C.; Rodríguez, R.M. Crosstalk Between Hypoxia and ER Stress Response: A Key Regulator of Macrophage Polarization. Front. Immunol. 2019, 10, 2951.
- Labiano, S.; Palazon, A.; Melero, I. Immune Response Regulation in the Tumor Microenvironment by Hypoxia. Semin. Oncol. 2015, 42, 378–386.
- Kawanaka, T.; Kubo, A.; Ikushima, H.; Sano, T.; Takegawa, Y.; Nishitani, H. Prognostic significance of HIF-2alpha expression on tumor infiltrating macrophages in patients with uterine cervical cancer undergoing radiotherapy. J. Med. Investig. 2008, 55, 78–86.
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421.
- Imtiyaz, H.Z.; Williams, E.P.; Hickey, M.M.; Patel, S.A.; Durham, A.C.; Yuan, L.-J.; Hammond, R.; Gimotty, P.A.; Keith, B.; Simon, M.C. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. J. Clin. Investig. 2010, 120, 2699–2714.
- Burke, B.; Giannoudis, A.; Corke, K.P.; Gill, D.; Wells, M.; Ziegler-Heitbrock, L.; Lewis, C.E. Hypoxia-induced gene expression in human macrophages: Implications for ischemic tissues and hypoxia-regulated gene therapy. Am. J. Pathol. 2003, 163, 1233–1243.
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e813.
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage Expression of Hypoxia-Inducible Factor-1α Suppresses T-Cell Function and Promotes Tumor Progression. Cancer Res. 2010, 70, 7465–7475.
- Ohta, A.; Diwanji, R.; Kini, R.; Subramanian, M.; Ohta, A.; Sitkovsky, M. In vivo T Cell Activation in Lymphoid Tissues is Inhibited in the Oxygen-Poor Microenvironment. Front. Immunol. 2011, 2.
- Thiel, M.; Caldwell, C.C.; Kreth, S.; Kuboki, S.; Chen, P.; Smith, P.; Ohta, A.; Lentsch, A.B.; Lukashev, D.; Sitkovsky, M.V. Targeted Deletion of HIF-1α Gene in T Cells Prevents their Inhibition in Hypoxic Inflamed Tissues and Improves Septic Mice Survival. PLoS ONE 2007, 2, e853.
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019, 458, 13–20.
- Messai, Y.; Gad, S.; Noman, M.Z.; Le Teuff, G.; Couve, S.; Janji, B.; Kammerer, S.F.; Rioux-Leclerc, N.; Hasmim, M.; Ferlicot, S.; et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel-Lindau Gene Mutation Status. Eur. Urol. 2016, 70, 623–632.
- Pouyssegur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443.
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241.
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794.
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577.
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819.
- Nakagawa, Y.; Negishi, Y.; Shimizu, M.; Takahashi, M.; Ichikawa, M.; Takahashi, H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol. Lett. 2015, 167, 72–86.
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9.
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137.
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. ImmunoTher. Cancer 2018, 6, 57.
- Casanello, P.; Torres, A.; Sanhueza, F.; Gonzalez, M.; Farias, M.; Gallardo, V.; Pastor-Anglada, M.; San Martin, R.; Sobrevia, L. Equilibrative nucleoside transporter 1 expression is downregulated by hypoxia in human umbilical vein endothelium. Circ. Res. 2005, 97, 16–24.
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 42, 101304.
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10.
- Mancino, A.; Schioppa, T.; Larghi, P.; Pasqualini, F.; Nebuloni, M.; Chen, I.-H.; Sozzani, S.; Austyn, J.M.; Mantovani, A.; Sica, A. Divergent effects of hypoxia on dendritic cell functions. Blood 2008, 112, 3723–3734.
- Zhu, Y.; Huang, B.; Shi, J. Fas ligand and lytic granule differentially control cytotoxic dynamics of natural killer cell against cancer target. Oncotarget 2016, 7, 47163–47172.
- Barsoum, I.B.; Hamilton, T.K.; Li, X.; Cotechini, T.; Miles, E.A.; Siemens, D.R.; Graham, C.H. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: Role of nitric oxide. Cancer Res. 2011, 71, 7433–7441.
- Balsamo, M.; Manzini, C.; Pietra, G.; Raggi, F.; Blengio, F.; Mingari, M.C.; Varesio, L.; Moretta, L.; Bosco, M.C.; Vitale, M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur. J. Immunol. 2013, 43, 2756–2764.
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486.
- Martinez-Zaguilan, R.; Seftor, E.A.; Seftor, R.E.; Chu, Y.W.; Gillies, R.J.; Hendrix, M.J. Acidic pH enhances the invasive behavior of human melanoma cells. Clin. Exp. Metastasis 1996, 14, 176–186.
- Zhang, J.; Han, C.; Dai, H.; Hou, J.; Dong, Y.; Cui, X.; Xu, L.; Zhang, M.; Xia, Q. Hypoxia-Inducible Factor-2α Limits Natural Killer T Cell Cytotoxicity in Renal Ischemia/Reperfusion Injury. JASN 2016, 27, 92–106.
- Keck, M.K.; Zuo, Z.; Khattri, A.; Stricker, T.P.; Brown, C.D.; Imanguli, M.; Rieke, D.; Endhardt, K.; Fang, P.; Brägelmann, J.; et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV subtypes. Clin. Cancer Res. 2015, 21, 870–881.
- Duechler, M.; Peczek, L.; Zuk, K.; Zalesna, I.; Jeziorski, A.; Czyz, M. The heterogeneous immune microenvironment in breast cancer is affected by hypoxia-related genes. Immunobiology 2014, 219, 158–165.
- Deng, B.; Zhu, J.-M.; Wang, Y.; Liu, T.-T.; Ding, Y.-B.; Xiao, W.-M.; Lu, G.-T.; Bo, P.; Shen, X.-Z. Intratumor Hypoxia Promotes Immune Tolerance by Inducing Regulatory T Cells via TGF-β1 in Gastric Cancer. PLoS ONE 2013, 8, e63777.
- Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Ioannou, K.; Ziogas, A.C.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. Br. J. Cancer 2012, 107, 1869–1875.
- Chouaib, S.; Messai, Y.; Couve, S.; Escudier, B.; Hasmim, M.; Noman, M.Z. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front. Immunol. 2012, 3, 21.
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44.
This entry is offline, you can click here to edit this entry!