Taken with the growing importance of cathepsin-mediated substrate proteolysis in tumor biology and progression, the focus and emphasis placed on therapeutic design and development is coming into fruition. Underpinning this approach is the invariable progression from the direction of fully characterizing cathepsin protease members and their substrate targets, towards targeting such an interaction with tangible therapeutics. The two groups of such substrates that have gained much attention over the years are the pro- and anti- apoptotic protein intermediates from the extrinsic and intrinsic signaling arms of the apoptosis pathway.
- apoptosis
- Bcl-2
- BH3
- extrinsic
- intrinsic
- MOMP
- cell death
- cancer
- cathepsins
- catheptasome
1. Introduction
Over the last 10 years, great strides have been taken in identifying cathepsin protease-specific substrates, which have revealed a number of interesting regulatory paradigms based on the growing pleiotropic nature of these enzymes. While they were originally isolated as lysosomal proteases involved in the proteolysis of intracellular proteins, their role in determining the fate of cells as active components arising from the lysosomal ‘suicide bag’ have seen their importance evolve [1,2]. At the molecular level, mechanistic insights into their substrate specificity have been slow to take shape as the cathepsin family of proteases is composed of a large number of members and subgroups, and which have similar yet distinct biochemical properties and substrate specificity [3,4]. Briefly, this protease family contains 15 members that can be further sub-divided into aspartic (D and E), serine (A and G), or cysteine cathepsin proteases (B, C, F, H, K, L, O, R, S, V, X/Z), and endo- and exo-peptidases [5]. Herein, the cysteine cathepsin proteases have received considerable attention based on their ability to remain catalytically active at relatively low and neutral pH [6,7]. Mechanistically, cathepsin proteases have been reported as tightly regulated at the protein level by their cognate inhibitors, the cystatins, but have also been reported as deregulated and overexpressed in a number of disease states such as cancer, and thus have a high level of therapeutic, diagnostic, and prognostic value [8]. Moreover, they are emerging to possess diversity in their subcellular compartmentalization, based on them reported to reside cytoplasmically, within the nucleus, with mitochondria, in addition to being localized within the extracellular compartment [5,9].
When taken with the ability of cathepsins to reside in the cytoplasm, the potential for them to cleave and modulate a number of biochemically significant signaling pathway intermediates central to determining cell viability has taken on heightened importance. One such process that the cathepsins have been identified to regulate is apoptosis (reviewed in [5,10]). While apoptosis was originally discovered as an important mechanism in cell fate and tissue development, it has emerged over time as a central regulatory mechanism in the development and progression of certain cancers [11], autoimmune diseases [12], and neurodegenerative disorders [13,14].
Generally, apoptosis is the end-point of two main regulatory pathways [15]. Firstly, the extrinsic pathway links extracellular death-inducing signals through receptor engagement, which culminates in caspase activation. For example, the binding of Fas or TNF-alpha ligands to their cognate receptors permit the formation of a death inducing signaling complex (DISC) and which signals through the activation of the cysteine-aspartic proteases caspase-8 or -10, to activate effector caspase-3, which can cleave a number of cellular macromolecules that initiate apoptosis [16]. Secondly, the intrinsic pathway, which is mainly regulated by the mitochondrion in response to cellular stress or growth factor deprivation [17,18], is responsible for the release of a number of mitochondrial-derived pro-apoptotic proteins. Here, this key regulatory step of mitochondrial outer membrane permeabilization (MOMP) can be mechanistically induced by a number of pro-apoptotic proteins from the B-cell lymphoma (Bcl-2) family, which mediate the formation of the mitochondrial permeability transition pore and have been the basis of many excellent studies that highlight significant value in therapeutically targeting this step [19]. The subsequently released mitochondrial proteins include Smac/DIABLO, apoptosis inducing factor (AIF), and cytochrome c [20,21], and form what is referred to as the ‘apoptosome complex’, which gives rise to the activation of caspases-9 and -3 [22,23,24]. The activation of the execution caspases -3, -6, and -7, leads to the activation of cytoplasmic CAD nucleases, which degrade nuclear lamin proteins, inhibit the DNA repair enzyme PARP, and which gives rise to the variety of morphological changes that are characteristic of apoptosis [25]. Lastly, externalization of phosphatidylserine at the apoptotic cell surface signals the recognition of apoptotic cells and their uptake by phagocytosis [26].
Throughout this process, and of central importance from a regulatory standpoint, is the expression of the Bcl-2 family of proteins [27], the members of which determine the sensitivity of cells to apoptosis through the intrinsic apoptosis pathway. Broadly, these proteins can be divided into two main functional groups: the pro-apoptotic and the anti-apoptotic proteins, and both of which constitute a family of proteins that are in excess of 18 members [28]. Alternatively, this Bcl-2 family can be subdivided structurally into three main groups, based upon the similarities between their Bcl-2 Homology (BH) domains [29,30]. Firstly, the pro-apoptotic protein group includes BAX, Bcl-2-associated protein X; BAK, Bcl-2 antagonist/killer; and BOK, Bcl-2 ovarian killer, and all structurally contain the four BH 1–4 domains and directly promote MOMP [31,32,33]. Secondly, members from the anti-apoptotic protein group all contain the four BH 1–4 domains: Bcl-2, B cell lymphoma-2; Bcl-xL, Bcl-2-related protein X; Bcl-w; Mcl-1; A1; or Bcl-B, which suppress MOMP by binding BAK or BAX directly. Thirdly, the pro-apoptotic BH3-only proteins group include Bim, Bcl-2 interacting mediator of cell death; Bad, Bcl-2 antagonist of cell death; BID, Bcl-2 interacting domain death agonist; Bmf, Bcl-2 modifying factor; Bik, Bcl-2 interacting killer-like protein; Noxa; Puma, p53-upregulated modulator of apoptosis and Hrk (Harakiri). As seen from Figure 1, all such members (except BID) contain one highly conserved BH3 domain, which is present in most pro-apoptotic proteins and may not always be absent in anti-apoptotic proteins [34]. However, the BH4 motif is present in all Bcl-2 anti-apoptotic proteins as a conserved domain [31,35].
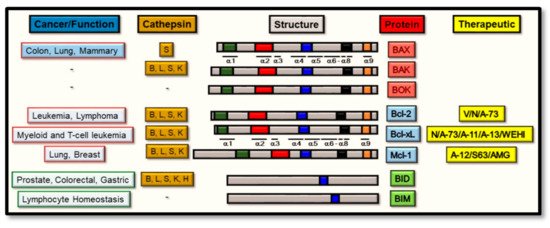
Therapeutically, both extrinsic and intrinsic pathway intermediates have been the subject of intense scrutiny from a therapeutic targeting standpoint, helped and aided by the elucidation of the crystal structure of many signaling intermediates derived from these pathways [36]. Clearly, the rationale underlying most recent therapeutic development strategies have been directed at selectively manipulating the two arms of the apoptotic pathway with a view to harnessing the cells own molecular signaling machinery to mediate cell death [37], through a number of approaches including the design of ‘BH3-mimetics’ [38,39,40]. Functionally, mutagenesis of the BH3 domain (spanning α-helices 1–2, [41]) from activated BAX (or BAK) was unveiled to highlight its critical homo-oligomerization role during the induction of apoptosis [42]. As a domain that also binds the hydrophobic groove of the anti-apoptotic proteins (spanning α-helices 2–5, [43]), this interaction gives effect to the inhibitory properties of anti-apoptotic proteins such as Bcl-xL [44], thus preventing BAX activation and homo-oligomerization. Consequently, targeting this interaction site between BAX (or BAK) and the anti-apoptotic Bcl-2 protein, through the design of BH3-mimetics was unveiled to hold high therapeutic potential [45]. While this has some clear-cut relevance in cancer development and progression (where deregulated apoptotic pathway intermediates commonly prevail [46]), a complex picture is, however, emerging about how the proposed therapeutic modulation of these pathways may also be interconnected with other signaling cascades of relevance. This is of particular importance for the avoidance of therapeutic-mediated side effects [47].
Mechanistically, the cathepsin proteases have been linked to regulating both of the intrinsic and extrinsic signaling cascades, and which forms the basis of this review article by addressing what recent evidence supports their input into this regulatory step of apoptosis. Herein, we outline the rationale for therapeutically targeting certain cathepsin proteases, in the context of either selectively abrogating their activity for the breakdown (and destabilization of the pro-apoptotic Bcl-2 proteins), or their activity to be selectively maintained for the breakdown of the anti-apoptotic Bcl-2 proteins. Clearly, a favorable endpoint is effectively altering the balance of active pro-apoptotic Bcl-2 proteins levels in relation to their anti-apoptotic counterparts, with a view to tipping the balance of these proteins, in order to favor apoptosis in the context of killing cancer cells and halting tumor progression (Figure 2). Finally, we discuss what potential exists in furthering such findings to incorporate the simultaneous co-modulation of cathepsin protease activity and its relationship shared with pro- and anti- apoptotic Bcl-2 protein members, with a view to highlighting how the therapeutic design of BH3-mimetic can be utilized for greater effect.
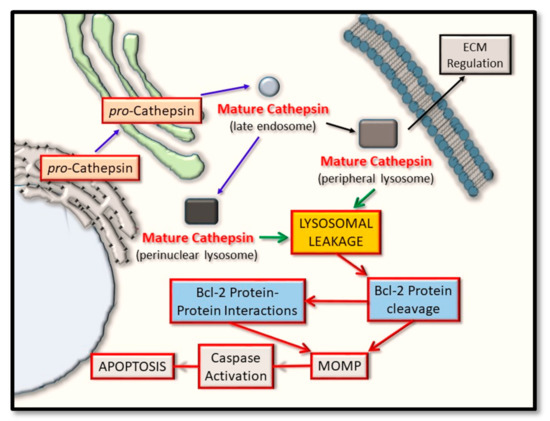
Figure 2. Cathepsin protein trafficking and intrinsic pathway activation for apoptosis. Cathepsin proteases are synthesized in their pro-inactive forms and mature through pro-domain removal, as they take on an endosomal and lysosomal localization. Through lysosomal leakage, they are released into the cytoplasm where they have the potential to proteolytically cleave certain Bcl-2 family protein members and thus alter their ability to form oligomers. Such events converge on regulating mitochondrial outer membrane permeabilization (MOMP) and have the effect of regulating caspase activation and cellular apoptosis.
2. Anti-Apoptotic Bcl-2 Proteins as Substrates for the Cathepsin Proteases
The anti-apoptotic Bcl-2 proteins have also gained increasing importance for therapeutic targeting, from their abilities to drive cancer progression and the most extensive studies of which have described the targeting of Bcl-2, Bcl-xL, and Mcl-1 [81,82]. Mechanistically, while cancer cells can contain an abundance of pro-apoptotic Bcl-2 proteins, the amplification of anti-apoptotic Bcl-2 proteins can lead to BH3-only proteins being competitively depleted [83,84]. In the instance of the Bcl-2 anti-apoptosis protein, its amplification and overexpression can give rise to a number of hematological malignancies and solid tumors, such as lymphoma, prostate cancer, and small cell lung cancer [85,86,87]. Similarly, Bcl-xL can also drive tumor progression in the presence of Bcl-2 protein-directed therapeutic resistance [88,89,90], while Mcl-1 amplification has been reported in lung and breast cancers [88] and, thus, carries equal levels of importance. In all of these instances, the over-expression of these anti-apoptotic proteins have the effect of damaged cells surviving longer, thus permitting the accumulation of additional genetic lesions that can contribute to driving tumor progression [91,92]. In support, mouse knockout studies have yielded invaluable insights through delineating the dispensability of some anti-apoptotic proteins in cancer progression. For example, in Bcl-2−/− mice, abnormal death of lymphocytes had been reported [91], while Bcl-xL−/− mice possessed abnormalities in the demise of neurons and erythroid progenitor cells [92]. Additionally, Mcl-1−/− mouse studies reported the death of early stage embryos and mice with conditional deletions experienced a rapid loss of mature lymphocytes or hematopoietic stem cells [93,94].
Based on the above evidence, the consequential role played by the cathepsin proteases during anti-apoptotic protein proteolysis has taken on significant importance. This was addressed by Droga-Mazovec et al. (2008), who reported the cleavage of proteins Bcl-2, Bcl-xL, and Mcl-1 upon the treatment of a variety of cell lines with the lysosomorphic agent, LeuLeuOMe [78]. More specifically, cathepsins -B, -L, -S, and -K were observed to cleave Bcl-2, Bcl-xL, and Mcl-1 proteins, using purified recombinant proteins in an in vitro cleavage assay at pH 7.2. As such, for substrates that are anti-apoptotic regulators, potential inhibition of cognate cathepsin proteases overexpressed during cancer development, can predictably have the effect of enhancing the levels of certain anti-apoptotic Bcl-2 sub-family members, and thus drive cancer progression. However, the significant (and simultaneous) benefits here may stem from preventing the proteolysis of pro-apoptotic Bcl-2 family members (such as BAX and BAK), particularly by cathepsins -B, -L, -K, and -S. Therefore, an ideal solution might involve the design of therapeutics that take on the properties of BH-3 mimetics, but have the flexibility and selectively, to inhibit cathepsin proteases against either of their pro-apoptotic or the anti-apoptotic protein substrates.
As very promising candidates for targeted cancer therapy, early studies identifying novel therapeutics targeting the Bcl-2 subfamily of anti-apoptosis proteins involved the screening of natural compounds and which yielded little success [95]. Through structural studies and rational drug-design approaches, a number of BH3-mimetics have been identified, and which promisingly act through binding the hydrophobic groove of the anti-apoptotic Bcl-2 protein, thus permitting enhanced monomeric BAK and BAX proteins to become activated through chemotherapeutic stimulation [83,96]. Such an approach led to the design of ABT-263 (or Navitoclax), a small inhibitor directed at Bcl-2, Bcl-xL, and BCl-w [84,97]. While it showed promising efficacy during phase I-II clinical trials for treating B-cell malignancies [97], its use was limited due to it inducing platelet-depleting effects [98,99,100,101]. Later, the cause of this was reported to be due to Bcl-xL inhibition having the effect of negatively modulating circulating platelets [102,103]. Nevertheless, a derivative of ABT-263 (called Venetoclax) did offer a successful treatment for patients with chronic lymphocytic leukemia (CLL), elapsed or refractory CLL and acute myeloid leukemia (AML) [104,105,106]. While this agent was largely ineffective for most solid tumors and chemotherapeutic resistance was common [107], it was evaluated as a useful therapeutic for treating thrombocytopenia [89]. This also formed the basis for the development of derivatives such as WEHI-539, A1155463, and A-1331852 [108,109,110], and from which, A-1331852 was reported as the first successful antagonist for Bcl-xL targeting. In the context of Mcl-1 inhibition, ABT-737, Venetoclax (or Navitoclax) [111,112] were evaluated as being largely ineffective therapeutics, but nevertheless highlights the specificity and exclusivity with which they can target certain anti-apoptotic targets [113,114]. Collectively, while such approaches do indeed highlight the power of BH3-mimetics, as with most therapeutics, their further development may be a necessity as therapeutic resistance and side effects can still present significant hurdles.
As seen from our recent studies outlining an alternative approach for developing a peptide inhibitor to target cathepsin S-specific Bcl-2 family intermediates of the intrinsic pathway, effective peptide efficacy (and specificity) may also be guided by the intracellular pH of cancer cells, as a key-determining factor. In this context, we observed that cathepsin S could cleave Bcl-xL in vitro better at pH 7 than at pH 5 and that this reaction could be inhibited better at pH 7 (than at pH 5), by the novel peptide inhibitor CS-PEP1. Such a pH-sensitive cleavage reaction appeared fortuitously to favor the inhibition of Bcl-xL cleavage (by CS-PEP1) at a relatively higher pH, in relation to the lower pH that can be prevalent during tumor development and apoptosis. Favorably, cathepsin S-mediated cleavage of BAX (and the inhibition of this reaction by CS-PEP1) was observed to be effective at both pH 5 and 7 [79]. Collectively, such findings offer an alternative approach in targeting this inhibitory axis of the intrinsic pathway for apoptosis with greater flexibility, through designing a more selective therapeutic that is based upon combining the principles of BH3-mimetics with the classical approach of cathepsin S-directed allosteric inhibition [79]. Importantly, the inhibition of cathepsin-mediated BAX (or possibly BAK) and Bcl-xL cleavage, may also be modulated further in a pH-dependent manner by such a therapeutic, thus permitting the favorable cleavage of an anti-apoptotic protein better than its pro-apoptotic counterpart.
3. Cathepsin and the Bcl-2 Proteins: Targeted Therapeutic Development
While a number of validated therapeutics directed at the Bcl-2 anti-apoptotic proteins have been described herein, further questions do arise to address what effects such therapeutics may have on the activity of upstream modulators, such as the cathepsin proteases. For example, during the inhibition of BAX-Bcl-2 protein binding, are BAX (or Bcl-2) protein molecules presented as good substrates for their cognate cathepsin proteases in the presence of BH3-mimetics? Based on recent developments (reported herein), this could indeed have an impact on the overall net stability of certain Bcl-2 proteins and how well they may fulfill their native roles as key apoptotic regulators.
As a novel and alternative ‘semi-rational’ approach for therapeutic development, we have defined a peptide therapeutic based on its ability to potentially disrupt the BAX BH3-Bcl-xL hydrophobic groove interaction, while at the same time taking on properties of an allosteric inhibitor directed at the catalytic activity of cathepsin S. Of relevance, may also be the subcellular compartmentalization shared by the cathepsin and Bcl-2 proteins, and the penetrability of any arising therapeutics. In this context, while targeting the lysosome may be achieved with relative ease, other extra-lysosomal compartments, such as the unidentified and membranous perinuclear compartment in which enriched cathepsin S and BAX-derived proteins have been uniquely seen to co-localize (and for which we refer to as the ‘catheptasome’, until further characterization) may present additional challenges [79]. This is a key factor in any therapeutic targeting strategy and is one that warrants further consideration, in this instance. Nevertheless, all of the above approaches do indeed offer an alternative to therapeutic design and assessment, through their potential to target the regulatory effects of the cathepsin proteases as upstream regulators of the Bcl-2 family of proteins (Figure 3).
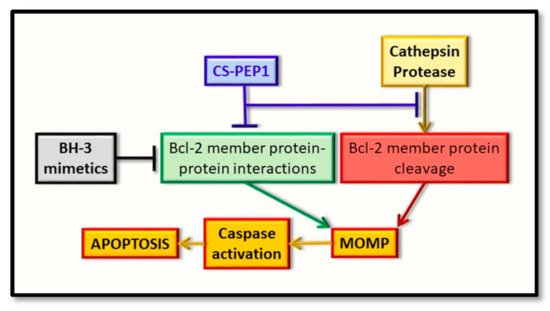
Figure 3. Representation of the early intrinsic arm of the apoptotic pathway, highlighting the key points identified for therapeutic intervention. Whereas BH3-mimetics (black box) can interfere with the pro-apoptotic BH3-domain interaction with the hydrophobic groove of the anti-apoptotic protein (green box), the novel inhibitor (CS-PEP1, blue box) can interfere with cathepsin S-mediated cleavage of the BAX protein (red box) while also potentially interfering with the respective BH3 domain-hydrophobic groove of BAX with Bcl-xL (green box). Consequently, BAX protein can be stabilized in a manner where it can be readily activated for the induction of MOMP and thus enhance apoptosis (orange boxes).
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094669