Advances achieved with molecular biology and genomics technologies have permitted investigators to discover epigenetic mechanisms, such as DNA methylation and histone posttranslational modifications, which are critical for gene expression in almost all tissues and in brain health and disease. These advances have influenced much interest in understanding the dysregulation of epigenetic mechanisms in neurodegenerative disorders. Although these disorders diverge in their fundamental causes and pathophysiology, several involve the dysregulation of histone methylation-mediated gene expression. Interestingly, epigenetic remodeling via histone methylation in specific brain regions has been suggested to play a critical function in the neurobiology of psychiatric disorders, including that related to neurodegenerative diseases. Prominently, epigenetic dysregulation currently brings considerable interest as an essential player in neurodegenerative disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), Amyotrophic lateral sclerosis (ALS) and drugs of abuse, including alcohol abuse disorder, where it may facilitate connections between genetic and environmental risk factors or directly influence disease-specific pathological factors.
- epigenetics
- Alzheimer’s disease
- Parkinson’s disease
- Huntington’s disease
- Amyotrophic lateral sclerosis
- neuronal loss and alcohol
1. Introduction
Neurodegenerative (ND) disorders are among the leading bases of disability and death worldwide [1,2,3]. The neurodegeneration process involves progressive atrophy of neurons, leading to loss of neuronal connectivity and function followed by its demise, thereby adversely affecting brain function. Despite decades of basic and clinical research, most strategies designed to reverse degenerative brain diseases are analgesic. This is not surprising as neurodegeneration progresses quietly for decades before the appearance of symptoms. Most important advances in sequencing technologies have allowed the mapping of transcriptomic patterns in human postmortem brain tissues in various ND disorders, including in vitro and in vivo cell and animal models. These investigations facilitated the discovery of classical neurodegeneration pathways and uncovered novel targets, including synaptic degeneration in the majority of ND, that share several puzzling characteristics. For example, although large patient populations’ intense genetic evaluation has been performed, a substantial proportion of ND incidents have no known genetic origin [4], and only a few neuro-pathophysiology studies have identified gene mutations or defective genes. The majority of ND studies recognized the contribution of adverse environmental conditions, such as exposure to toxins, chemicals, nutritional deficits, social factors, drug abuse, and alcohol, leading to neurodegeneration and manifestation of pathology and behavioral defects. Many medications and therapies have been evaluated for these diseases, resulting in less than acceptable results [5,6]. Therefore, the necessity for novel treatments to improve symptoms and prevent ND progression is at an all-time high.
2. PTM of DNA-Associated Histone Proteins
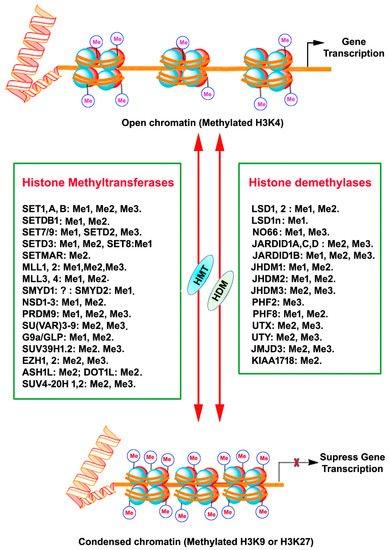
3. Histone Methylation
Histone methylation usually occurs at the arginine or lysine N-terminal region, which leads to the activation or suppression of gene expression. Each arginine residue in histones can be subjected to monomethylation and symmetric/asymmetric dimethylation. Likewise, each lysine residue can be mono-, di- and tri-methylated, and also nearby residues can form different methylation combinations [20,21,22,23]. Methylation of all the arginine residue is catalyzed by the family of arginine methyltransferase (PRMT) enzymes. PRMT1 monomethylates and asymmetrically dimethylates arginine residues, thus resulting in the activation of gene expression. PRMT2 promotes gene suppression through monomethylation and symmetric dimethylation of arginine residues. Moreover, the cofactor-associated arginine methyltransferase (CARM1) promotes gene activation through monomethylation and asymmetric dimethylation of arginine residues. HMTs that catalyze the methylation of lysine amino acids of histone proteins and other proteins are called lysine methyltransferases (KMTs) [24] for their extensive enzymatic activity and precise substrate specificity (Figure 1). All KMTs, except for KMT4, have a conserved Su (var)3–9, Enhancer of Zeste, Trithorax (SET) domain that presents catalytical activity [25]. The presence of a characteristic homologous sequence [24,26] has enabled the classification KMTs into well-defined subcategories. The KMTs that add the methyl group on H3K9, H3K27, and H4K20 are the main repressive KMTs, while the KMTs that add methyl group on H3K4, H3K14, and H3K36 are classified as activating KMTs [27]. One of the most highly investigated marks is H3 lysine (4) (H3K4) methylation, which is associated with active gene expression. Histone H3K4 methylation is catalyzed by a group (KMT2) of mixed lineage leukemia (MLL) proteins (SET1A, SET1B, MLL1, MLL2, MLL3, MLL4, and ASH1) [28,29,30]. H3K4me3 mark is found mainly in nucleosomes associated with the promoter regions of actively transcribed genes [31], while H3K4me2 is located in the gene bodies and enhancers associated with active genes [18]. H3K4me1 is found in enhancers, promoters, and at the 3′ end of active genes [32]. A family of methyltransferases (KMT1) (SUV39H1, SUV39H2, G9A/GLP and ESET/SETDB1; KMT8: RIZ1) catalyze histone H3K9 methylation and catalytical activity differ with catalyzing substrates and resulting products. Histone H3K9 trimethylation (H3K9me3) is catalyzed by SUV39 and results in a heterochromatin structure and transcriptional suppression [33]. Histone H3K9 dimethylation (H3K9me2) is catalyzed by G9A and results in a euchromatin structure and suppressed gene expression [34,35]. G9A-like protein (GLP) forms a hetero polypeptide complex with G9A, and, collectively, they catalyze the H3K9 dimethylation [36]. ESET/SETDB1 catalyzes the trimethylation of H3K9 [37], which results in the inhibition of gene expression. Among the alterations linked with transcription elongation, histone H3K36 trimethylation modification occurring at nucleosomes in the 3′ region of the transcription region of active genes [38]. H3K36me3 is catalyzed by KMT3 (SET2, SYMD2 and NSD1) enzymes [39,40,41]. Histone H3K79 di- and tri-methylation are catalyzed by DOT1L enzymes (KMT4) and are associated with gene expression activation [42]. Histone H4K20 mono- or tri-methylation is catalyzed by SET8 and SUV420H1/2 enzymes (KMT5), respectively, associated with gene inactivation [43]. The monomethylation of H3K27 often increases the expression of target genes, whereas the addition of three methyl groups at the same site usually suppresses gene transcription [44]. Histone H3K27me1 is catalyzed by ATXR5 and ATXR6 or TXR1 [45,46] and H3K27me3 is established by Enhancer of Zeste 2 (EZH2) of the polycomb repressive complex 2 (PRC2) [47,48,49].
4. Alzheimer’s Disease (AD)
5. Huntington’s Disease (HD)
6. Parkinson’s Disease (PD)
PD is the second most predominant neurodegenerative disorder in the world after AD [136]. The most significant pathology of PD is the demise of dopaminergic neurons in the substantia nigra with Lewy bodies (aggregated alpha-synuclein and ubiquitin-protein and damaged nerve cells as cytoplasmic inclusions) [137,138,139]. PD is characterized by motor symptoms (bradykinesia, tremor, postural instability, and rigidity) and nonmotor symptoms (cognitive issues, autonomic dysfunction, and REM sleep behavior disorder) [137]. However, the mechanisms involved in PD pathogenesis have not been fully identified. The specific loss of dopaminergic neurons in PD has been believed to be a consequence of complex interactions between genetic and environmental factors. Nevertheless, the characteristics of the relationship between the two significant changes remain to be established. A growing body of studies have begun to support epigenetic events, such as DNA methylation and histone modification in PD progression [140,141,142].
A mounting number of studies overwhelmingly suggest the critical function of the histone modification and PD-associated α-synuclein coding gene SNCA expression [143], meaning that histone methylation also may have a crucial role in the regulation of the SNCA gene. Interestingly, overexpression of α-SYN in flies and neuronal cells, such as SH-SY5Y enhanced G9a, H3K9me1, and H3K9me2 levels and H3K9me2-target genes (L1cam, Snap25) eventually lead to impaired synaptic activity [144]. In contrast, H3K4me3 was significantly enriched at the SNCA promoter region in postmortem brain samples from patients with PD and matched controls [145]. Similarly, using dead Cas9-Suntag system-mediated locus-specific approaches, the reduction in H3K4me3 from the SNCA promoter reduced α-synuclein levels in neuronal cell lines and PD-derived induced pluripotent stem cell lines (iPSCs) [145]. Significant H3K4me3 enrichment was observed at the SNCA promoter in the neuronal nuclei (NeuN) of positive neurons of substantia nigra (SN) tissue samples from PD patients. Interestingly, significant reductions in H3K4me3 and H3K27me3 marks were observed in SH–SY5Y cells treated with the neurotoxin 6-hydroxydopamine (6-OHDA) [146]. In the same model, pretreatment with GSK-J4, a potent inhibitor of KDM6A/B and KDM5B/C (demethylates H3K27me3/me2 and H3K4me3/me2 respectively) [147,148], significantly prevented H3K4me3 and H3K27me3 marks’ decrease [146]. In contrast to the cell culture model, increased H3K27me3 levels were found in PD patients’ brains [145], indicating the possible role of PRC2 in vivo PD models. These findings indicate that abnormal histone methylation, such as H3K4me3 (gene activation) and H3K27me3 (gene suppression), control gene expression (e.g., α-synuclein) in SN neurons from patients with PD. Because specific histone methylation is one of the central regulators of gene expression, future investigation is warranted to understand further how specific histone methylation of the gene s, including the SNCA gene, directly regulates the accessibility of transcription factors that can access gene regulatory regions. Finally, there are still undiscovered histone methylation marks that may regulate synaptic and pathological abnormalities found in PD. Those may also have a considerable impact on regulating chromatin structure and overall effects on gene transcription in PD and should be of future interest.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094654