Mitochondrial dysfunction results in a series of defective cellular events, including decreased adenosine triphosphate (ATP) production, enhanced reactive oxygen species (ROS) output, and altered proteastasis and cellular quality control. An enhanced output of ROS may damage mitochondrial components, such as mitochondrial DNA and elements of the electron transport chain, resulting in the loss of proper electrochemical gradient across the mitochondrial inner membrane and an ensuing shutdown of mitochondrial energy production. Neurons have an increased demand for ATP and oxygen, and thus are more prone to damage induced by mitochondrial dysfunction. Mitochondrial dysfunction, damaged electron transport chains, altered membrane permeability and Ca2+ homeostasis, and impaired mitochondrial defense systems induced by oxidative stress, are pathological changes involved in neurodegenerative disorders. A growing body of evidence suggests that the use of antioxidants could stabilize mitochondria and thus may be suitable for preventing neuronal loss. Numerous natural products exhibit the potential to counter oxidative stress and mitochondrial dysfunction; however, science is still looking for a breakthrough in the treatment of neurodegenerative disorders. β-caryophyllene is a bicyclic sesquiterpene, and an active principle of essential oils derived from a large number of spices and food plants. As a selective cannabinoid receptor 2 (CB2) agonist, several studies have reported it as possessing numerous pharmacological activities such as antibacterial (e.g., Helicobacter pylori), antioxidant, anti-inflammatory, analgesic (e.g., neuropathic pain), anti-neurodegenerative and anticancer properties. The present review mainly focuses on the potential of β-caryophyllene in reducing oxidative stress and mitochondrial dysfunction, and its possible links with neuroprotection.
- oxidative stress
- mitochondrial dysfunction
- neurodegeneration
- β-caryophyllene
- neuroprotection
1. Introduction
Mitochondrial dysfunction refers to an impairment in mitochondrial function, resulting in a series of defective cellular events including decreased adenosine triphosphate (ATP) production, enhanced reactive oxygen species (ROS) output, altered proteostasis and cellular quality control [1]. Neurons have increased demand for ATP and oxygen, with cortical neurons being known to consume approximately 4.7 billion ATP molecules per second, and thus they are more prone to the damage induced by mitochondrial dysfunction [2]. Such critical requirements for ATP and oxygen make them susceptible to electron leakage from the electron transport chain, resulting in generation of free radicals and induced oxidative stress [3]. Furthermore, lowered levels of antioxidant defenses further increase the neuronal susceptibility to mitochondria induced oxidative damage [4]. In addition, a large ROS output not only damages the biomolecules of the neuronal cells, but also damages mitochondrial components (e.g., mitochondrial DNA) and elements of the electron transport chain, resulting in a loss of electrochemical gradient across the mitochondrial inner membrane and the ensuing shutdown of mitochondrial energy production [5].
The electron transport chain, or mitochondrial respiratory chain, consists of five complexes (complex I, II, III, IV and V) and is one of the major structural and functional components of mitochondria, catalyzing the phosphorylation of adenosine diphosphate (ADP) to ATP [6]. These complexes are comprised of over 80 proteins, 13 of which are encoded by mitochondrial DNA and are components of oxidative phosphorylation [6][7]. Complexes I–IV constitute the electron transport chain, which generates water by oxidation of hydrogen (derived from organic acids like pyruvic and fatty acids) with atomic oxygen [8]. ATP production involves two coordinated processes, including transport of electrons along the complexes to produce water and the pumping of protons across the mitochondrial inner membrane (from matrix to intermembrane space) through complexes I, III and IV. ATP is thus generated by the influx of these protons back to the matrix through complex V [9][10][11]. Under normal physiological conditions, 1.5% of the oxygen may be converted into ROS, which suggests that the majority of intracellular ROS is generated by mitochondria [12]. The production of superoxide and other reactive oxygen species occurs primarily at complexes I and III [13]. Under pathological conditions, the highly reactive hydroxyl ions could damage mitochondrial DNA, proteins, and lipids, resulting in the defective functioning of complexes I and III, causing superoxide radical formation by increased electron reduction of oxygen, leading to metabolic oxidative stress, genomic instability, and cellular injury [14][15][16][17].
Damaged mitochondrial DNA may decrease the expression of critical proteins of the electron transport chain, amplifying oxidative stress which eventually triggers apoptosis [14]. The electron transport chain is also sensitive to nitrosative stress, as nitration can modify mitochondrial proteins, causing alterations in the functioning of many metabolic enzymes in the electron transport chain, such as nicotinamide adenine dinucleotide (NAD) dehydrogenase, cytochrome c oxidase, and ATP synthase [18]. Most importantly, acute exposure to ROS inactivates the iron-sulfur centers of complexes I, II and III, while chronic exposure can damage cellular and mitochondrial proteins, lipids, and genetic materials [7]. ROS also alters mitochondrial membrane permeability, as the inner membrane is located near the site of ROS production and thus is more prone to lipid peroxidation [19]. Peroxidation of mitochondrial phospholipids may increase the proton permeability of the inner membrane, which under normal physiological conditions is permeable only to tiny neutral molecules [20]. Increased membrane permeability could lead to altered fluidity, as well as impaired biochemical functions of numerous transporters and enzymes present in mitochondrial membranes [12].
Mitochondria play a critical role in regulating neuronal Ca2+ homeostasis, and genetic and pharmacologic manipulations enhancing mitochondrial Ca2+ sequestration may protect neuronal cells against excitotoxicity [21]. Excessive ROS generation alters mitochondrial Ca2+ homeostasis, where peroxynitrite inactivates key mitochondrial enzymes, affecting the energy status of the cell and triggering the release of Ca2+ from the mitochondria [22]. Elevated Ca2+ levels cause a shift in mitochondrial potential and result in the production of superoxide radicals which may lead to a vicious cycle. Changes in mitochondrial permeability in Ca2+ overloaded mitochondria result in osmotic swelling and the rupture of the outer mitochondrial membrane [23]. ROS production in mitochondria further promotes Ca2+ uptake and enhances membrane permeability, and eventually results in the release of cytochrome c and the initiation of apoptosis [24]. Figure 1 depicts the links between oxidative stress and mitochondrial dysfunction, and their possible impact on aging and disease development and progression.
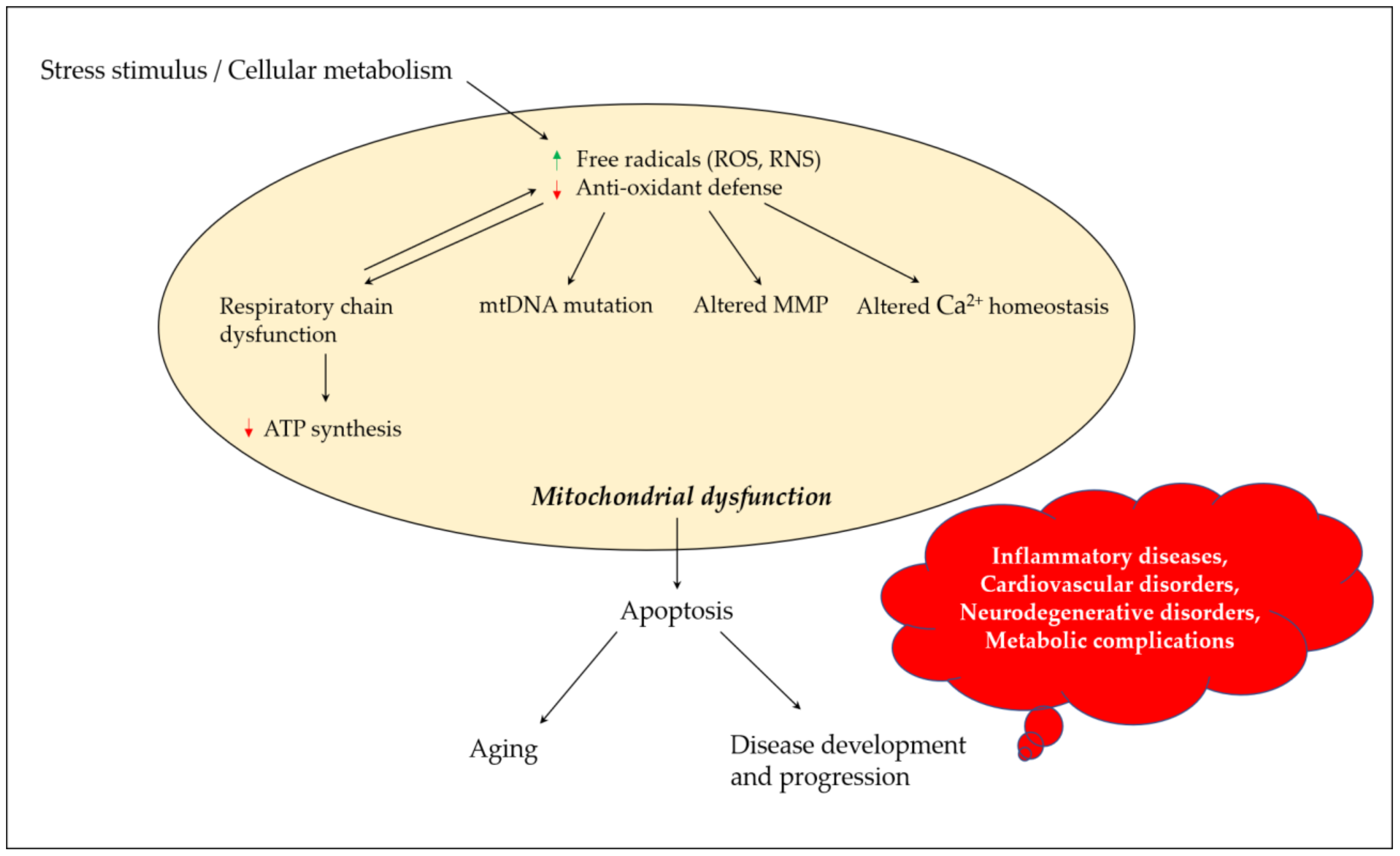
Figure 1. Mitochondrial dysfunction and its contribution towards aging and disease development and progression. Stress stimulus and irregular cellular metabolism may lead to the increased production of ROS and RNS, and decreased antioxidant defense parameters, which eventually result in mitochondrial dysfunction due to a defective mitochondrial respiratory chain, mutation in mtDNA, altered MMP and influenced Ca2+ homeostasis. These events could promote apoptosis, paving the road for aging and disease development and progression. ROS, reactive oxygen species; RNS, reactive nitrogen species; mtDNA, mitochondrial DNA; MMP, mitochondrial membrane potential; ATP, adenosine triphosphate.
Many antioxidant agents were found to be active when tested in animal models of neurodegeneration (e.g., vitamin E), but unfortunately showed no or little benefits in clinical settings. Identifying bioactive substances that can counter oxidative damage as well as restore mitochondrial dysfunction may be a fruitful approach in reversing neurodegeneration [3].
2. β-Caryophyllene: Alteration of Oxidative Stress and Mitochondrial Dysfunction
A number of studies (Table 1) have suggested the alteration of oxidative stress and mitochondrial dysfunction by β-caryophyllene and β-caryophyllene-containing vegetable extracts, as one of the potential mechanisms in protecting neurons from degeneration [25]. Chávez-Hurtado et al. (2020) observed a reduction in DNA oxidation and overexpression of glial fibrillary acidic proteins with β-caryophyllene (10 mg/kg, p.o. for 4 weeks) in the prefrontal cortex and hippocampus of BALB/c mice withd-galactose induced aging [26]. In an in vivo model of PD, β-caryophyllene (50 mg/kg, i.p. for 4 weeks) ameliorated oxidative stress (restored antioxidant enzymes, increased GSH, and inhibited lipid peroxidation), neuroinflammation (decreased levels of IL-1β, IL-6, and TNF-α, and downregulated COX-2 and iNOS expression), and glial activation as well as rescuing dopaminergic neurons [27].
Table 1. Alteration of oxidative stress and mitochondrial dysfunction by β-caryophyllene and vegetable extracts containing β-caryophyllene.
| Study Model | Extract or Compound (Dose/Concentration) |
Study Outcomes | References |
|---|---|---|---|
| BALB/c mice, with d-galactose induced aging | β-caryophyllene (10 mg/kg/day, p.o. for 4 weeks) |
↓ DNA oxidation and overexpression of glial fibrillary acidic proteins in the prefrontal cortex and hippocampus. | [26] |
| Rats, with PD | β-caryophyllene (50 mg/kg/day, i.p. for 4 weeks) |
↑ GSH, SOD and CAT. Inhibit lipid peroxidation. ↓ IL-1β, IL-6, and TNF-α levels. ↓ COX-2 and iNOS expression. ↓ glial activation and rescued dopaminergic neurons. |
[27] |
| Rats with PD | β-caryophyllene (50 mg/kg/day, i.p.) for 4 weeks |
↓ pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α) and inflammatory mediators (NF-κB, COX-2, and iNOS). ↑ glutathione, SOD and CAT. ↓ lipid peroxidation. |
[28] |
| C6 glioma cell line | β-caryophyllene (0.5 and 1.0 μM) |
↑ cellular antioxidant responses via CB2 receptor dependent Nrf2 activation. ↓ ROS production. Restored MMP. |
[29] |
| Neurovascular unit (BMECs, neurons and astrocytes) | β-caryophyllene (10 μmol/L) |
↓ BBB permeability, neuronal apoptosis, oxidative stress damage, inflammatory cytokines. ↓ metalloproteinase-9 expression/activity, Bax expression. ↑ expression of claudin-5, occludin, ZO-1, GAP-43, and Bcl-2. |
[30] |
| Adult male Sprague–Dawley rats, with focal cerebral ischemia | β-caryophyllene (34, 102 and 306 mg/kg/day, p.o.). |
↑ Nrf2 and HO-1 expression. Restored SOD and CAT activity and expression. |
[31] |
| C57BL/6 mice, with autoimmune encephalomyelitis | β-caryophyllene (25 and 50 mg/kg/day, p.o.). |
↓ H2O2, IFN-γ, TNF-α, IL-17 and NO. | [32] |
| Human neuroblastoma SH-SY5Y cells | β-caryophyllene (1 and 2.5 μM) |
Restored reduction in MMP. ↑ intracellular GSH and GPx activity. ↓ Caspase-3 and Bax. Restored Bcl-2 expression Suppressed HO-1 activation and JNK phosphorylation. |
[33] |
| Wistar rats-male with Aβ (1-42)-induced memory impairment | Pinus halepensis essential oil (1 and 3%). | ↓ hippocampal AChE activity. ↑ hippocampal antioxidant markers (SOD, CAT, GPx and GSH). ↓ malondialdehyde (MDA) levels. |
[34] |
| CAD neuroblastoma cell lines | Aloysia citrodora Palau essential oil (0.01 and 0.001 mg/mL) | ↓ H2O2 (250 μM) and Aβ (10 μM) induced neurotoxicity. Fe2+ chelation in vitro. |
[35] |
| Rats, with ICV colchicine induced memory impairment | Syzygium aromaticum (L.) Merr. and L.M. Perry (0.05 mL/kg and 0.1 mL/kg) | ↓ AChE activity, lipid peroxidation levels, and nitrite concentrations. Restored activities of GSH and mitochondrial respiratory enzyme complex (I–IV). |
[36] |
| Male mice, with PD | Eplingiella fruticosa leaf essential oil. (5 mg/kg/day, p.o. for 40 days) |
↓ membrane lipid peroxide levels in the striatum. ↑ dopamine levels in the striatum and substantia nigra pars compacta. |
[37] |
| Male adult Wistar albino rats, with induced MS like manifestations | Ocimum basilicum L. essential oil (100 and 200 μL/kg) | ↓ proinflammatory cytokines (TNF-α and IL-6) in prefrontal cortex. ↓ astrogliosis by increasing GFAP and Iba-1 levels in prefrontal cortex. ↑ mitochondrial function, integrity, respiratory control rate and ATP production. ↓ mitochondria-dependent apoptosis in prefrontal cortex of rats. |
[38] |
| In vitro, antioxidant and AChE inhibition assays | Salvia rosmarinus Spenn. essential oil. | Strong antioxidant effects (DPPH, ABTS, FRAP and β-carotene bleaching tests). Significant AChE inhibition. |
[39] |
AChE, acetylcholinesterase; Bax, Bcl-2-associated X protein; BBB, blood–brain barrier; Bcl-2, B-cell lymphoma 2; BMECs, brain microvascular endothelial cells; CAT, catalase; CB2 receptor, cannabinoid-2 receptor; GAP-43, growth-associated protein-43; GFAP, Glial fibrillary acidic protein; GPx, glutathione peroxidase; GSH, glutathione; H2O2, hydrogen peroxide; HO-1, heme oxygenase-1; Iba-1, ionized calcium binding adaptor molecule-1; IL-1β, interleukin-1β; IL-6, interleukin-6; iNOS, inducible nitric oxide synthase; JNK, c-Jun N-terminal kinase; MDA, malondialdehyde; MMP, mitochondrial membrane potential; Nrf2, nuclear factor erythroid 2–related factor 2; ROS, reactive oxygen species; SOD, superoxide dismutase; TNF-α, tumor necrosis factor- α; ZO-1, zonula occludens-1; “↓” reduction; “↑”increment.
3. Concluding Remarks
Oxidative stress and mitochondrial dysfunction are well organized events in the degeneration of neuronal cells. Targeting mitochondria in chronic degenerative disorders is not a novel idea. Many promising experimental therapeutics are known to enhance mitochondrial function, unfortunately none have yet proven successful in halting the development and progression of neurodegeneration. Literature data suggest that β-caryophyllene, a dietary phytocannabinoid, possesses a neuroprotective capability through decreasing oxidative stress and stabilizing mitochondria, and could be a potential lead molecule in the discovery of drugs for neurodegenerative disorders. Besides CB2 receptor agonism, β-caryophyllene has been found to positively regulate PPAR-γ, TLRs and neuroimmune pathways, as possible targets implicated in the protection against neuronal loss. Essential oils containing β-caryophyllene, extracted from different vegetable sources, also showed promising neuroprotective effects following their attenuation of oxidative stress and/or mitochondrial dysfunction. However, it remained unknown whether these beneficial effects could be attributed to the summation of the activities of phytoconstituents present in essential oils, or to a single compound mediating the observed effects. Nevertheless, the available data are not sufficient to draw any clinical conclusion for the recommendation of β-caryophyllene in the management of neurodegenerative disorders, and an expansion of the literature is strongly needed, in particular regarding the most effective doses for beneficial roles of β-caryophyllene in the management of neurodegenerative disorders, and the potential benefits of β-caryophyllene in targeting mitochondria in neurodegenerative diseases utilizing both experimental and human studies.
This entry is adapted from the peer-reviewed paper 10.3390/antiox10040546
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159.
- Islam, M.T. Oxidative Stress and Mitochondrial Dysfunction-Linked Neurodegenerative Disorders. Neurol. Res. 2017, 39, 73–82.
- Facecchia, K.; Fochesato, L.A.; Ray, S.D.; Stohs, S.J.; Pandey, S. Oxidative Toxicity in Neurodegenerative Diseases: Role of Mitochondrial Dysfunction and Therapeutic Strategies. J. Toxicol. 2011, 2011, 683728.
- Kumar, A. Mitochondrial Dysfunction & Neurological Disorders. Curr. Neuropharmacol. 2016, 14, 565.
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS Generation by Mitochondria in Cells with Impaired Electron Transport Chain and Mitochondrial DNA Damage. Mitochondrion 2007, 7, 106–118.
- Chen, J.Q.; Yager, J.D.; Russo, J. Regulation of Mitochondrial Respiratory Chain Structure and Function by Estrogens/Estrogen Receptors and Potential Physiological/Pathophysiological Implications. Biochim. Biophys. Acta Mol. Cell Res. 2005, 1746, 1–17.
- Ghezzi, D.; Zeviani, M. Assembly Factors of Human Mitochondrial Respiratory Chain Complexes: Physiology and Pathophysiology. Adv. Exp. Med. Biol. 2012, 748, 65–106.
- Shoubridge, E.A.; Wai, T. Mitochondrial DNA and the Mammalian Oocyte. Curr. Top. Dev. Biol. 2007, 77, 87–111.
- Sas, K.; Robotka, H.; Toldi, J.; Vécsei, L. Mitochondria, Metabolic Disturbances, Oxidative Stress and the Kynurenine System, with Focus on Neurodegenerative Disorders. J. Neurol. Sci. 2007, 257, 221–239.
- Porter, R.K.; Brand, M.D. Mitochondrial Proton Conductance and H+/O Ratio Are Independent of Electron Transport Rate in Isolated Hepatocytes. Biochem. J. 1995, 310, 379–382.
- Zhang, Y.; Wang, M.; Li, H.; Zhang, H.; Shi, Y.; Wei, F.; Liu, D.; Liu, K.; Chen, D. Accumulation of Nuclear and Mitochondrial DNA Damage in the Frontal Cortex Cells of Patients with HIV-Associated Neurocognitive Disorders. Brain Res. 2012, 1458, 1–11.
- Wei, Y.H.; Lu, C.Y.; Wei, C.Y.; Ma, Y.S.; Lee, H.C. Oxidative Stress in Human Aging and Mitochondrial Disease-Consequences of Defective Mitochondrial Respiration and Impaired Antioxidant Enzyme System. Chin. J. Physiol. 2001, 44, 1–11.
- Hollensworth, S.B.; Shen, C.C.; Sim, J.E.; Spitz, D.R.; Wilson, G.L.; Ledoux, S.P. Glial Cell Type-Specific Responses to Menadione-Induced Oxidative Stress. Free Radic. Biol. Med. 2000, 28, 1161–1174.
- van Houten, B.; Woshner, V.; Santos, J.H. Role of Mitochondrial DNA in Toxic Responses to Oxidative Stress. DNA Repair 2006, 5, 145–152.
- Voets, A.M.; Huigsloot, M.; Lindsey, P.J.; Leenders, A.M.; Koopman, W.J.H.; Willems, P.H.G.M.; Rodenburg, R.J.; Smeitink, J.A.M.; Smeets, H.J.M. Transcriptional Changes in OXPHOS Complex I Deficiency Are Related to Anti-Oxidant Pathways and Could Explain the Disturbed Calcium Homeostasis. Biochim. Biophys. Acta Mol. Basis. Dis. 2012, 1822, 1161–1168.
- Castro, M.D.R.; Castro, M.D.R.; Suarez, E.; Kraiselburd, E.; Isidro, A.; Paz, J.; Ferder, L.; Ayala-Torres, S. Aging Increases Mitochondrial DNA Damage and Oxidative Stress in Liver of Rhesus Monkeys. Exp. Gerontol. 2012, 47, 29–37.
- Alexeyev, M.F. Is There More to Aging than Mitochondrial DNA and Reactive Oxygen Species? FEBS J. 2009, 276, 5768–5787.
- Andreazza, A.C.; Shoo, L.; Wang, J.F.; Trevor Young, L. Mitochondrial Complex I Activity and Oxidative Damage to Mitochondrial Proteins in the Prefrontal Cortex of Patients with Bipolar Disorder. Arch. Gen. Psychiatry 2010, 67, 360–368.
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c Association with the Inner Mitochondrial Membrane Is Impaired in the CNS of G93A-SOD1 Mice. J. Neurosci. 2005, 25, 164–172.
- Stewart, V.C.; Heales, S.J.R. Nitric Oxide-Induced Mitochondrial Dysfunction: Implications for Neurodegeneration. Free Radic. Biol. Med. 2003, 34, 287–303.
- Mattson, M.R. Calcium and Neurodegeneration. Aging Cell 2007, 6, 337–350.
- Douarre, C.; Sourbier, C.; Dalla Rosa, I.; Brata Das, B.; Redon, C.E.; Zhang, H.; Neckers, L.; Pommier, Y. Mitochondrial Topoisomerase I Is Critical for Mitochondrial Integrity and Cellular Energy Metabolism. PLoS ONE 2012, 7, e41094.
- Joshi, G.; Sultana, R.; Perluigi, M.; Butterfield, D.A. In Vivo Protection of Synaptosomes from Oxidative Stress Mediated by Fe2+/H2O2 or 2,2 Azobis-(2-Amidinopropane) Dihydrochloride by the Glutathione Mimetic Tricyclodecan-9-Yl Xanthogenate. Free Radic. Biol. Med. 2005, 38, 1023–1031.
- Ross, W.N. Understanding Calcium Waves and Sparks in Central Neurons. Nat. Rev. Neurosci. 2012, 13, 157–168.
- Machado, K.C.; Islam, M.T.; Ali, E.S.; Rouf, R.; Uddin, S.J.; Dev, S.; Shilpi, J.A.; Shill, M.C.; Reza, H.M.; Das, A.K.; et al. A Systematic Review on the Neuroprotective Perspectives of Beta-Caryophyllene. Phytother. Res. 2018, 32, 2376–2388.
- Chávez-Hurtado, P.; González-Castañeda, R.E.; Beas-Zarate, C.; Flores-Soto, M.E.; Viveros-Paredes, J.M. β-Caryophyllene Reduces DNA Oxidation and the Overexpression of Glial Fibrillary Acidic Protein in the Prefrontal Cortex and Hippocampus of d-Galactose-Induced Aged BALB/c Mice. J. Med. Food 2020, 23, 515–522.
- Ojha, S.; Javed, H.; Azimullah, S.; Haque, M.E. β-Caryophyllene, a Phytocannabinoid Attenuates Oxidative Stress, Neuroinflammation, Glial Activation, and Salvages Dopaminergic Neurons in a Rat Model of Parkinson Disease. Mol. Cell. Biochem. 2016, 418, 59–70.
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321.
- Assis, L.C.; Straliotto, M.R.; Engel, D.; Hort, M.A.; Dutra, R.C.; de Bem, A.F. β-Caryophyllene Protects the C6 Glioma Cells against Glutamate-Induced Excitotoxicity through the Nrf2 Pathway. Neuroscience 2014, 279, 220–231.
- Tian, X.; Peng, J.; Zhong, J.; Yang, M.; Pang, J.; Lou, J.; Li, M.; An, R.; Zhang, Q.; Xu, L.; et al. β-Caryophyllene Protects in Vitro Neurovascular Unit against Oxygen-Glucose Deprivation and Re-Oxygenation-Induced Injury. J. Neurochem. 2016, 39, 757–768.
- Lou, J.; Cao, G.; Li, R.; Liu, J.; Dong, Z.; Xu, L. β-Caryophyllene Attenuates Focal Cerebral Ischemia-Reperfusion Injury by Nrf2/HO-1 Pathway in Rats. Neurochem. Res. 2016, 41, 1291–1304.
- Fontes, L.B.A.; Dias, D.; dos, S.; Aarestrup, B.J.V.; Aarestrup, F.M.; Filho, A.A.D.S.; Corrêa, J.O.d.A. β-Caryophyllene Ameliorates the Development of Experimental Autoimmune Encephalomyelitis in C57BL/6 Mice. Biomed. Pharmacother. 2017, 91, 257–264.
- Wang, G.; Ma, W.; Du, J. β-Caryophyllene (BCP) Ameliorates MPP+ Induced Cytotoxicity. Biomed. Pharmacother. 2018, 103, 1086–1091.
- Postu, P.A.; Sadiki, F.Z.; el Idrissi, M.; Cioanca, O.; Trifan, A.; Hancianu, M.; Hritcu, L. Pinus Halepensis Essential Oil Attenuates the Toxic Alzheimer’s Amyloid Beta (1-42)-Induced Memory Impairment and Oxidative Stress in the Rat Hippocampus. Biomed. Pharmacother. 2019, 112, 108673.
- Abuhamdah, S.; Abuhamdah, R.; Howes, M.-J.R.; Al-Olimat, S.; Ennaceur, A.; Chazot, P.L. Pharmacological and Neuroprotective Profile of an Essential Oil Derived from Leaves of Aloysia Citrodora Palau. J. Pharm. Pharmacol. 2015, 67, 1306–1315.
- Kumar, A.; Aggrawal, A.; Pottabathini, R.; Singh, A. Possible Neuroprotective Mechanisms of Clove Oil against Icv-Colchicine Induced Cognitive Dysfunction. Pharmacol. Rep. 2016, 68, 764–772.
- Beserra-Filho, J.I.A.; de Macêdo, A.M.; Leão, A.H.F.F.; Bispo, J.M.M.; Santos, J.R.; de Oliveira-Melo, A.J.; Menezes, P.D.P.; Duarte, M.C.; de Souza Araújo, A.A.; Silva, R.H.; et al. Eplingiella Fruticosa Leaf Essential Oil Complexed with β-Cyclodextrin Produces a Superior Neuroprotective and Behavioral Profile in a Mice Model of Parkinson’s Disease. Food Chem. Toxicol. 2019, 124, 17–29.
- Garabadu, D.; Singh, D. Ocimum Basilicum Attenuates Ethidium Bromide-Induced Cognitive Deficits and Pre-Frontal Cortical Neuroinflammation, Astrogliosis and Mitochondrial Dysfunction in Rats. Metab. Brain Dis. 2020, 35, 483–495.
- Leporini, M.; Bonesi, M.; Loizzo, M.R.; Passalacqua, N.G.; Tundis, R. The Essential Oil of Salvia Rosmarinus Spenn. From Italy as a Source of Health-Promoting Compounds: Chemical Profile and Antioxidant and Cholinesterase Inhibitory Activity. Plants 2020, 9, 798.