Warfarin has been utilized for decades as an effective anticoagulant in patients with a history of strong risk factors for venous thromboembolism (VTE).
- warfarin
- direct oral anticoagulants
- vascular calcification
1. History of Warfarin
2. Mechanism of Action and Properties
Factors II, VII, IX, and X and proteins C and S are blood clotting factors synthesized as inactive precursors. They undergo a vitamin-K-dependent post-translational modification, which involves the gamma carboxylation of glutamic acid residues [9]. These gamma-carboxyglutamyl residues bind calcium ions and are necessary for interaction with platelets. The complex of clotting factors with calcium ions binds phospholipids on platelets. The reaction is catalyzed by gamma-glutamyl carboxylase and requires carbon dioxide, oxygen, and the reduced form of vitamin K [9]. The reduced vitamin K cofactor is converted to vitamin K epoxide during the reaction. Vitamin K is regenerated from the epoxide by vitamin K epoxide reductase (Figure 1). Warfarin acts by inhibiting vitamin K epoxide reductase (Figure 2). The four coagulation factors involved have differing half-lives, up to 60 h [9].
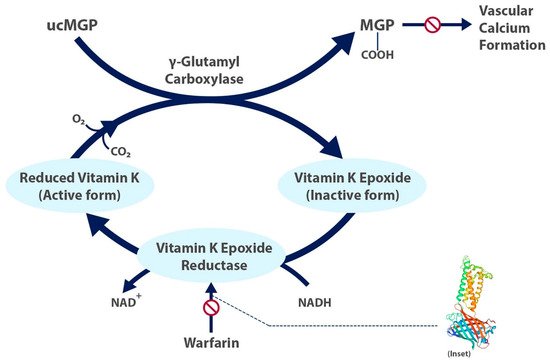
Factor VII has a half-life of 6 h, and therefore, warfarin begins to have an effect within approximately 24 h. Consequently, reversal following administration of vitamin K takes approximately 24 h. The peak effect may take up to 96 h. Protein C is a vitamin-K-dependent serine protease activated by thrombin. The activated form uses protein S as a cofactor and degrades factors Va and VIIIa. Both protein C and S are anticoagulant factors, and for this reason, a bridging agent, usually heparin, is necessary during a temporary hypercoagulable state that occurs before warfarin begins to take effect. Warfarin has no direct effect on an established thrombus, but once a thrombus has occurred, the goal of treatment is to prevent further enlargement of the formed clot. The anticoagulant effects of warfarin can be overcome by the administration of vitamin K.
The chemical properties of warfarin can influence metabolism and dosing in clinical practice. Warfarin is administered in the form of a racemic mixture of R and S enantiomers. The S-enantiomer is more active and possesses 2–5 times greater potency than the R-enantiomer [10,11]. Each isomer is metabolized distinctly in the liver. Oxidative metabolism of the S- isomer is affected by CYP2C9 [10]. The two most common genetic polymporphisms of CYP2C9 are CYP2C9*2 and CYP2C9*3 [11]. Multiple studies have confirmed that patients with these enzyme variants have reduced enzyme activity and require lower doses of warfarin to maintain a therapeutic INR [11].
This entry is adapted from the peer-reviewed paper 10.3390/cells10040773