Runx2 is essential for osteoblast differentiation, chondrocyte maturation, and transdifferentiation of terminally differentiated chondrocytes into osteoblasts. During osteoblast differentiation, Runx2 is weakly expressed in uncommitted mesenchymal cells, and its expression is upregulated in preosteoblasts, reaches the maximal level in immature osteoblasts, and is down-regulated in mature osteoblasts. Runx2 enhances the proliferation of osteoblast progenitors by directly regulating Fgfr2 and Fgfr3. Runx2 enhances the proliferation of suture mesenchymal cells and induces their commitment into osteoblast lineage cells through the direct regulation of hedgehog (Ihh, Gli1, and Ptch1), Fgf (Fgfr2 and Fgfr3), Wnt (Tcf7, Wnt10b, and Wnt1), and Pthlh (Pthr1) signaling pathway genes, and Dlx5. Runx2 heterozygous mutation causes open fontanelle and sutures because more than half of the Runx2 gene dosage is required for the induction of these genes in suture mesenchymal cells. Runx2 induces the proliferation of osteoblast progenitors and their differentiation into osteoblasts through reciprocal regulation via major signaling pathways, including Fgf, hedgehog, Wnt, and Pthlh, and transcription factors, including Sp7 and Dlx5. Runx2 also regulates the expression of bone matrix protein genes, including Col1a1, Col1a2, Spp1, and Bglap/Bglap2. Bglap/Bglap2 (osteocalcin) aligns biological apatite parallel to the collagen fibrils, which is important for bone strength, but osteocalcin does not play a role as a hormone in the pancreas, testis, and muscle.
- Runx2
- hedgehog
- Wnt
- Fgfr
- Pthr1
- Sp7
- proliferation
- differentiation
- cleidocranial dysplasia
- osteoblast
- osteocalcin
1. Introduction
Runx2 belongs to the Runx family, which has the DNA-binding domain runt, and consists of Runx1, Runx2, and Runx3 [1]. Runx2 heterodimerizes with Cbfb and acquires enhanced DNA binding ability and protein stability [2][3][4][5][6]. Runx2-deficient (Runx2–/–) mice lack osteoblasts and bone formation, and chondrocyte maturation is markedly inhibited [7][8][9][10]. In osteoblast differentiation, Runx2 is expressed in uncommitted mesenchymal cells, and its expression is upregulated in preosteoblasts, reaches the maximum level in immature osteoblasts, and is down-regulated in mature osteoblasts [11][12]. Runx2 regulates the proliferation of osteoblast progenitors, their commitment to osteoblast lineage cells, and the expression of bone matrix protein genes.
2. Reciprocal Regulation of the Essential Transcription Factors for Osteoblast Differentiation
Hedgehog signaling is essential for osteoblast differentiation in endochondral bone. Hedgehog binding to Ptch relieves the repression of Smo, which ultimately regulates Gli [13]. Hedgehog signaling is required for Runx2 expression in the perichondrium at the early process of osteoblast differentiation, which is the differentiation of osteoblast progenitors into preosteoblasts [14][15][16][17] (Figure 1). The sources of osteoblasts in primary spongiosa have been demonstrated to be Sp7-expressing perichondrial cells and the transdifferentiated osteoblasts from hypertrophic chondrocytes [18][19][20][21]. Runx2 is essential for the differentiation of perichondrial cells into osteoblasts and transdifferentiation of hypertrophic chondrocytes into osteoblasts [8][9][21]. Runx2 directly regulates Ihh expression in chondrocytes, osteoblast progenitors, and osteoblasts, as well as Gli1 and Ptch1expression in osteoblast progenitors and osteoblasts [12][22]. Thus, Runx2 and hedgehog signaling regulate each other, and induce osteoblast differentiation (Figure 1).
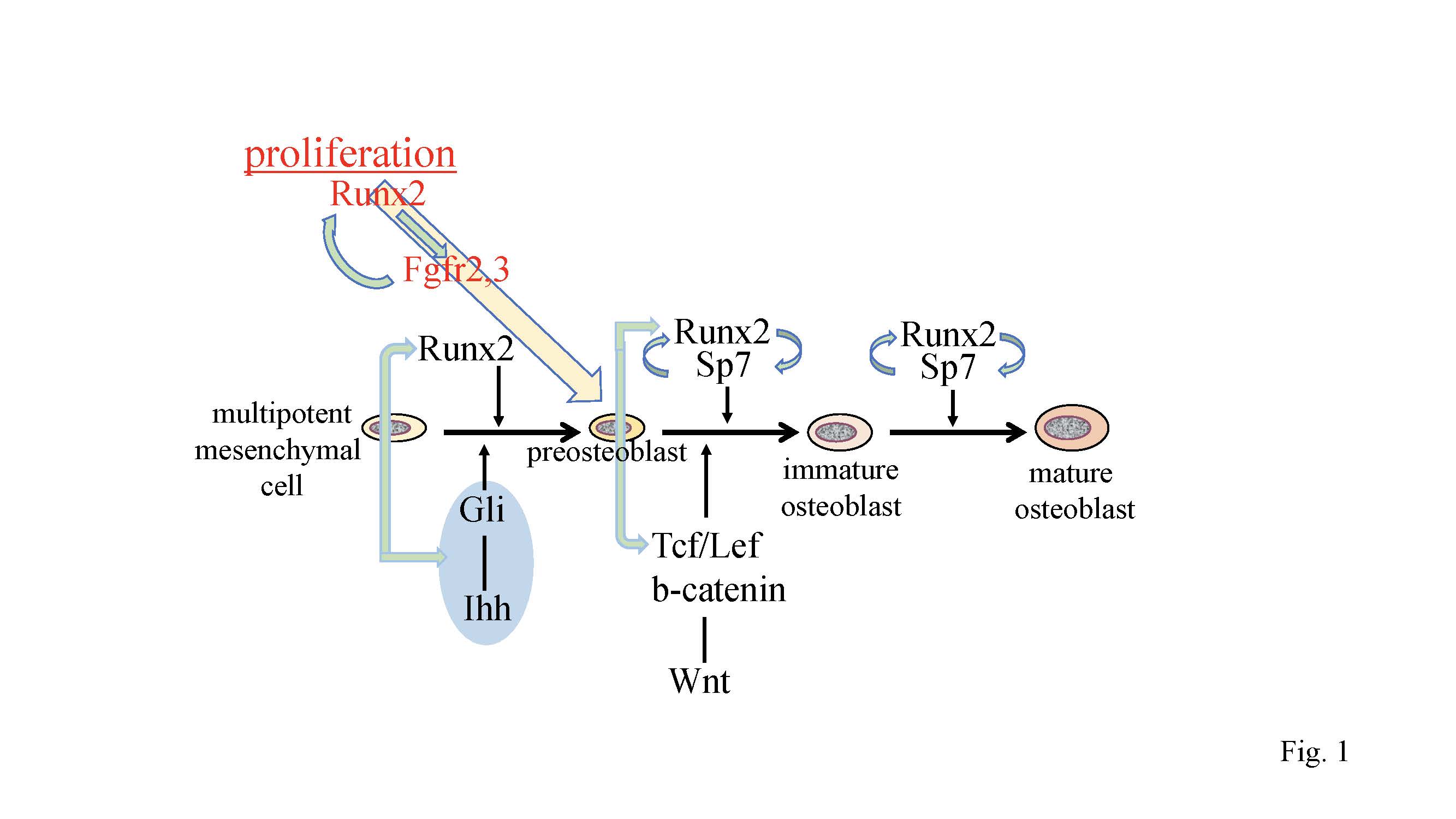
Figure 1. Regulation of osteoblast proliferation and differentiation by transcription factors. Runx2 induces the differentiation of multipotent mesenchymal cells into preosteoblasts. Ihh is required for the expression of Runx2 in the perichondrium of endochondral bones. Runx2 induces Sp7expression, and Runx2, Sp7, and canonical Wnt signaling induce the differentiation of preosteoblasts into immature osteoblasts. Runx2 and Sp7 are also involved in the maturation of osteoblasts. Runx2 regulates the proliferation of preosteoblasts by inducing Fgfr2 and Fgfr3. Runx2 expression and that of hedgehog, Fgf, and Wnt signaling pathway genes, and Sp7 are reciprocally regulated.
Runx2 directly regulates Sp7 expression, and osteoblasts and bone formation are also absent in Sp7–/– mice [23][24]. As Sp7 activates an osteoblast-specific enhancer of Runx2, Sp7 is also involved in the regulation of Runx2 expression [25] (Figure 1). Canonical Wnt signaling is also essential for osteoblast differentiation, because conditional Ctnnb1 knockout mice using Twist2 (Dermo1)-Cre, Col2a1-Cre, or Prrx1-Cre completely lack osteoblasts [26][27][28]. In these mice, Runx2 is expressed in the perichondrial cells, but Sp7 expression is weak or absent in perichondrial cells, indicating that osteoblast differentiation is arrested at the stage of preosteoblasts. Sp7–/– mice and conditional Ctnnb1–/– mice have abundant mesenchymal cells that express Runx2 in the presumptive bone region. As some of the mesenchymal cells in the perichondrium and calvaria differentiate into chondrocytes in Sp7–/– mice and conditional Ctnnb1–/– mice [26][27][28], Sp7 and canonical Wnt signaling inhibit chondrocyte differentiation and direct Runx2+ osteoblast progenitors to become osteoblasts (Figure 1).
3. Regulation of the Proliferation of Osteoblast Progenitors by Runx2
Overexpression of Runx2 under the control of the Prrx1 promoter accelerated osteoblast differentiation, inhibited chondrocyte differentiation, and caused limb defects [29]. The limb defects were caused by the ectopic induction of Fgfr1-3 by Runx2 [30]. The expression of Fgfr1-3 was directly regulated by Runx2, and Fgfr2 and Fgfr3 play roles in the proliferation of osteoblast progenitors. The expression of Fgfr1-3 was markedly reduced in Runx2–/– calvaria but not in Sp7–/– calvaria. Runx2 increased the proliferation of wild-type osteoblast progenitors and augmented Fgf2-induced proliferation. Thus, Fgf signaling plays a major role in the proliferation of osteoblast progenitors, and Runx2 regulates the proliferation of osteoblast progenitors by inducing Fgfr2 and Fgfr3 [30] (Figure 1).
It is difficult to investigate the proliferation of osteoblast lineage cells in vivo because osteoblast lineage cells at different stages of differentiation are mixed and the differentiation stage affects their proliferation. Both Runx2–/– mice and Sp7–/– mice have cartilaginous skeletons, and lack osteoblasts and bone formation [8][9][24]. Sp7–/– mice have abundant mesenchymal cells, which express Col1a1 weakly and are actively proliferating, in the presumptive bone regions, whereas Runx2–/– mice have few mesenchymal cells, which express Col1a1 at a markedly low level and have low proliferative activity, in the presumptive bone regions [30]. Furthermore, Runx2, Fgfr2, and Fgfr3 are expressed in the mesenchymal cells in Sp7–/– mice at levels comparable to those in osteoblasts in wild-type mice. These suggest that mesenchymal cells in Sp7–/– mice are preosteoblasts and that Sp7–/– mice are an appropriate model for the investigation of preosteoblast proliferation because osteoblast differentiation is blocked at the preosteoblast stage. Sp7–/– preosteoblasts proliferated at a similar level as wild-type osteoblast lineage cells in vivo, but they proliferated faster than wild-type osteoblast progenitors in vitro. Fgf2 augmented the proliferation of Sp7–/– preosteoblasts, whereas knockdown of Runx2 inhibited this augmentation and reduced the expression of Fgfr2 and Fgfr3. The amount and proliferation of preosteoblasts in Sp7–/– mice was halved in Sp7–/–Runx2+/– mice, indicating that preosteoblast proliferation is dependent on the gene dosage of Runx2. Therefore, Runx2 is required for preosteoblast proliferation in vivo, and Runx2 regulates it through the induction of Fgfr2 and Fgfr3 [30]. Moreover, Fgf2 enhances the Runx2 capacity for transcriptional activation via the PKC and MAPK pathways [30][31][32][33][34]. Thus, the Fgf signaling pathway and Runx2 positively regulate each other (Figure 1).
4. Molecular Mechanism of the Pathogenesis of Open Fontanelles and Sutures in Cleidocranial Dysplasia (CCD)
Although both intramembranous and endochondral bone development are affected in cleidocranial dysplasia (CCD), the open fontanelles and sutures and hypoplastic clavicles are typical features of CCD [12][35][36]. However, why the development of calvaria and clavicles is the most severely affected in CCD remains unclear.
In Runx2+/– mice, the closure of both posterior frontal (PF) and sagittal (SAG) sutures was interrupted. The suture mesenchymal cells expressed Sox9 at a similar level in wild-type and Runx2+/– mice. The suture mesenchymal cells also expressed Runx2, but the expression level in Runx2+/– mice was half of that in wild-type mice. The cell density and cell proliferation in Runx2+/– sutures was less than those in wild-type mice. The expression of hedgehog signaling genes (Gli1, Ptch1, and Ihh), Fgf signaling genes (Fgfr2 and Fgfr3), Wnt signaling genes (Tcf7 and Wnt10b), Pth1r, Dlx5, Tnc, and Ncam1 were less in PF and SAG sutures of Runx2+/– mice than in those of wild-type mice. Overexpression or knockdown of Runx2 and ChIP analysis demonstrated that these genes are directly regulated by Runx2 (Figure 2). However, the expression levels of these genes, except Dlx5, were similar in calvarial bone tissues between wild-type and Runx2+/– mice. Furthermore, osteoblast marker gene expression was not reduced in the calvarial bone tissues of Runx2+/– mice. These findings indicate that more than half of the Runx2 gene dosage is required for the expression of these Runx2 target genes in suture mesenchymal cells, but half of the Runx2 gene dosage is sufficient for it in differentiated osteoblasts [12].
In organ culture of Runx2+/– calvaria, the ligands or agonists for hedgehog, Fgf, Wnt, and Pthlh signaling pathways enhanced calvarial bone development and suture closure. Furthermore, the antagonists of hedgehog, Fgf, Wnt, and Pthlh signaling pathways inhibited calvarial bone development, suture closure, and proliferation of suture mesenchymal cells in the organ culture of wild-type calvaria. These findings suggested that hedgehog, Fgf, Wnt, and Pthlh signaling pathways are involved in the expansion, condensation, and commitment of suture mesenchymal cells to osteoblast lineage cells, and that Runx2 regulates these processes by inducing their signaling pathway genes [12] (Figure 2).
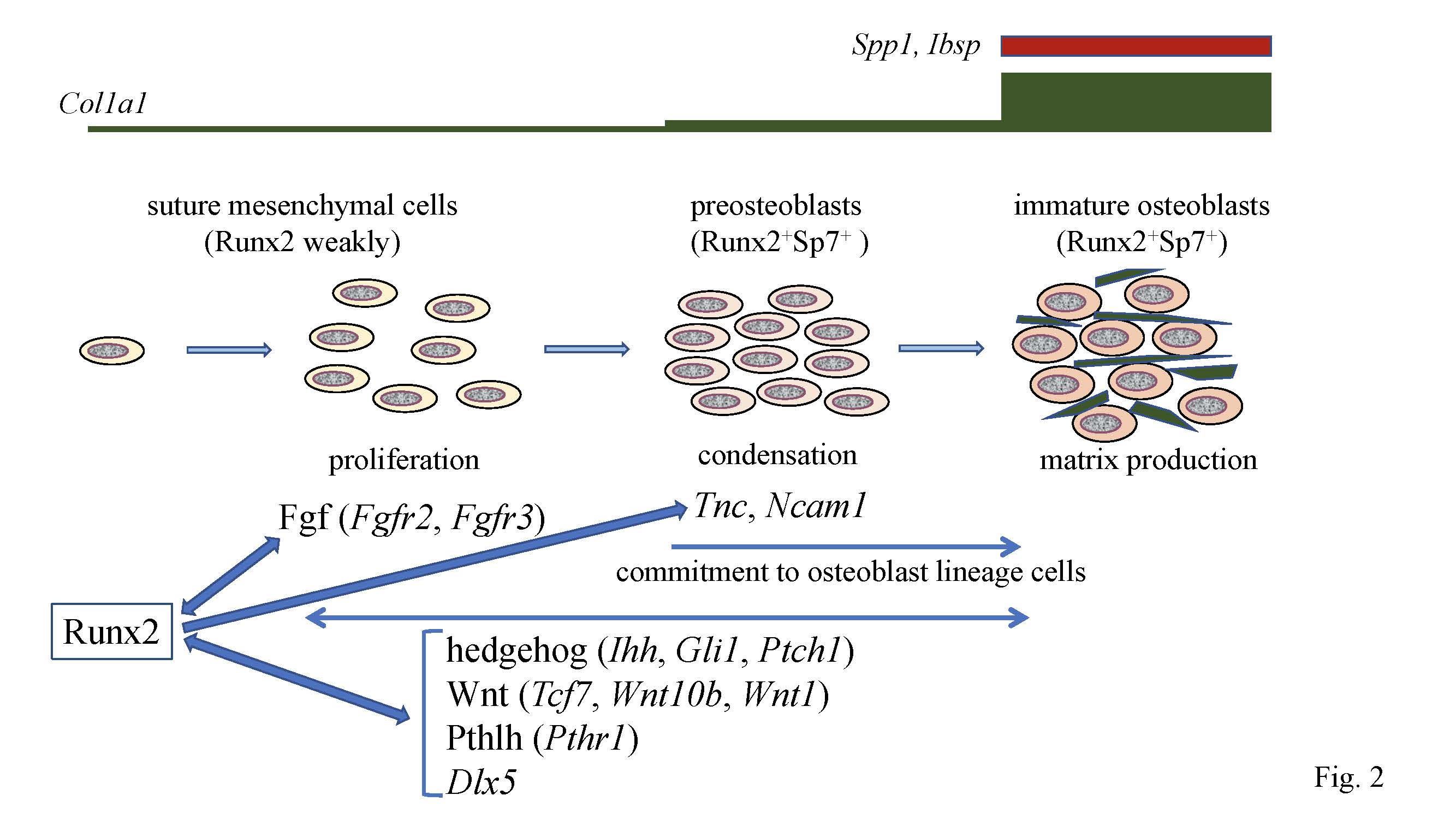
Figure 2. Calvarial bone development and suture closure. Suture mesenchymal cells weakly express Runx2, and its expression is upregulated in preosteoblasts and reaches the maximum level in immature osteoblasts. Runx2 induces Sp7 expression at the preosteoblast stage. Col1a1 expression is weak in suture mesenchymal cells, slightly upregulated in preosteoblasts, and markedly upregulated in immature osteoblasts, which also express Spp1and Ibsp. Runx2 increases the proliferation of suture mesenchymal cells and induces their commitment into osteoblast lineage cells through the induction of Fgf (Fgfr2 and Fgfr3), hedgehog (Ihh, Gli1, and Ptch1), Wnt (Tcf7, Wnt10b, and Wnt1), and Pthlh (Pthr1) signaling pathway genes, and Dlx5. Fgf signaling plays a role in proliferation, whereas the other genes function in both proliferation and commitment. There is reciprocal regulation between Runx2, and these signaling pathways and Dlx5. In the processes of commitment into osteoblast lineage cells, Runx2 also induces Tnc and Ncam1, which likely play roles in the condensation of suture mesenchymal cells, and these condensed mesenchymal cells then become preosteoblasts.
Runx2 directly regulates the expression of Tcf7, Wnt10b, and Wnt1, and Tcf7 and Ctnnb1 activate the P1 promoter and osteoblast-specific enhancer of Runx2 [12][30][37][38]. Thus, Wnt signaling and Runx2 mutually regulate their expression (Figure 1 and 2). As Dlx5 activates the P1 promoter and osteoblast-specific enhancer of Runx2 [30][39], Dlx5 and Runx2 also mutually regulate their expression (Figure 2). In addition, parathyroid hormone (PTH) increases Runx2 mRNA, Runx2 protein, and Runx2 activity, PTH induces Mmp13 promoter activity by activating Runx2 through PKA, and intermittent administration of PTH exerts lower anabolic effects on osteoblast-specific dominant-negative Runx2 or overexpressing Runx2 transgenic mice [11][40][41][42]. PTH and Pthlh share a common signaling pathway; therefore, there is also reciprocal regulation between the Pthlh signaling pathway and Runx2 (Figure 2).
5. The Functions of Runx2 in Bone Matrix Protein Gene Expression
After commitment to osteoblastic lineage cells, the osteoblasts express bone matrix protein genes at different levels depending on the maturational stage of the cells. Uncommitted mesenchymal cells weakly express Col1a1, its expression is slightly upregulated in preosteoblasts, and is markedly upregulated in immature osteoblasts [12] (Figure 2). Immature osteoblasts express Spp1 and then Ibsp, and mature osteoblasts strongly express Col1a1 and Bglap2 [11][43] (Figure 2).
Runx2–/– mice lack osteoblasts, and the expression of bone matrix protein genes, including Spp1, Ibsp, and Bglap/Bglap2, is absent and Col1a1expression is very low in the presumptive bone regions [7][8]. Although osteoblasts are observed in type II Runx2-specific knockout mice, the expression of Col1a1, Spp1, and Bglap/Bglap2 is reduced [44]. In vitro studies also demonstrated that Runx2 is a positive regulator that can upregulate the expression of bone matrix protein genes, including Col1a1, Spp1, Ibsp, Bglap/Bglap2, and Fn1 [45][46][47][48]. Moreover, reporter assays revealed that Runx2 activates the promoters of bone matrix protein genes, including Col1a1, Col1a2, Spp1, and Bglap/Bglap2 [46][47][49][50].
The functions of osteocalcin (Ocn), which is encoded by Bglap/Bglap2 that are directly regulated by Runx2, were controversial. Ocn was demonstrated to inhibit bone formation and function as a hormone, which regulates glucose metabolism in the pancreas, testosterone synthesis in the testis, and muscle mass, based on the phenotype of Ocn–/– mice by Karsenty’s group [51][52][53][54]. Recently, Ocn–/– mice were newly generated by two groups independently [55][56]. Bone strength is determined by bone quantity and quality. The new Ocn–/– mice revealed that Ocn is not involved in the regulation of bone formation and bone quantity, but that Ocn regulates bone quality by aligning biological apatite (BAp) parallel to the collagen fibrils [56]. Moreover, glucose metabolism, testosterone synthesis and spermatogenesis, and muscle mass were normal in the new Ocn–/– mice [55][56]. Thus, the function of Ocn is the adjustment of growth orientation of BAp parallel to the collagen fibrils, which is important for bone strength to the loading direction of the long bone. However, Ocn does not play a role as a hormone in the pancreas, testis, and muscle (Fig. 3) [57].
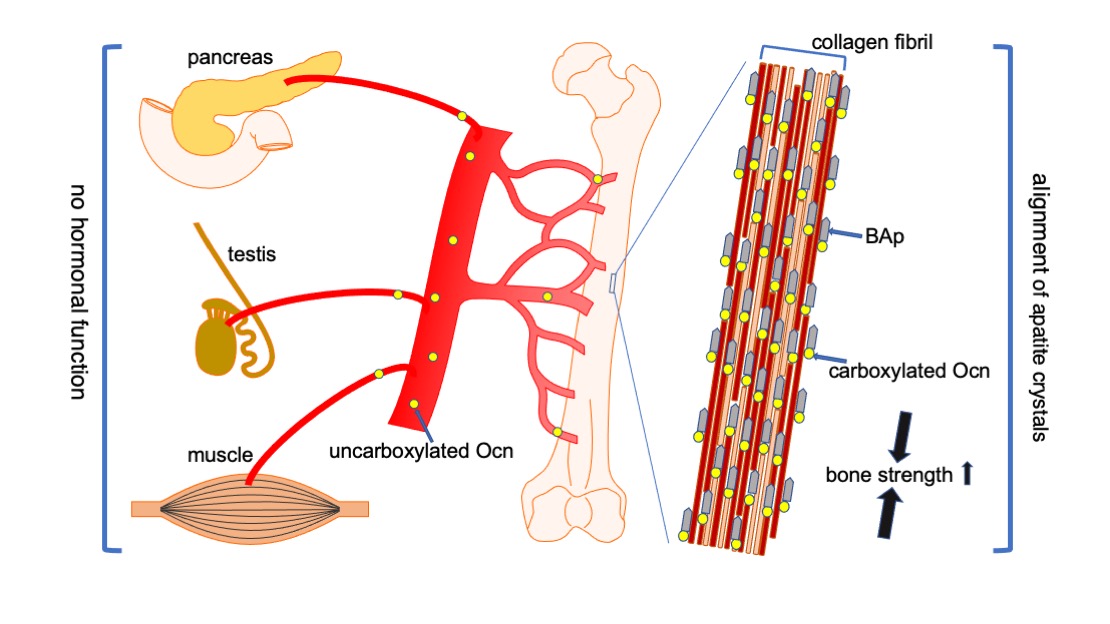
Figure 3. Functions of Ocn in bone, pancreas, testis, and muscle. Carboxylated Ocn is required for the alignment of BAp parallel to the collagen fibers and optimal bone strength. However, two newly generated Ocn–/– mouse lines and Ocn–/– rats [58] did not exhibit the impaired glucose metabolism, reduced testosterone synthesis and spermatogenesis, and reduced muscle mass observed in the Ocn–/– mouse line generated by Karsenty’s group. Thus, uncarboxylated Ocn does not physiologically function as a hormone that regulates glucose metabolism in the pancreas, testosterone synthesis in testis, or muscle mass.↑: Bone strength is increased by carboxylated Ocn regulates the alignment of .
6. Conclusions
Runx2 is required for the proliferation of preosteoblasts in whole skeletons and mesenchymal cells in sutures. Indeed, Runx2 is required for the commitment of mesenchymal cells to osteoblast lineage cells. Thus, Runx2 makes a condensed cell layer of uncommitted mesenchymal cells or osteoblast progenitors by increasing their proliferation and facilitates their differentiation into osteoblast lineage cells. Runx2 can exert multiple functions through reciprocal regulation via major signaling pathways, including Fgf, hedgehog, Wnt, and Pthlh, and transcription factors, including Sp7 and Dlx5. Osteoblast proliferation and differentiation are likely regulated by such reciprocal regulation rather than the cascade of transcription factors. Runx2 target protein Ocn regulates the alignment of BAp and is required for bone strength but not for glucose metabolism in the pancreas, testosterone synthesis in testis, or muscle mass.
This entry is adapted from the peer-reviewed paper 10.3390/ijms20071694
References
- Komori, T., Roles of Runx2 in Skeletal Development. Exp. Med. Biol. 2017, 962, 83–93.
- Kundu, M.; Javed, A.; Jeon, J.P.; Horner, A.; Shum, L.; Eckhaus, M.; Muenke, M.; Lian, J.B.; Yang, Y.; Nuckolls, G.H.; Stein, G.S.; Liu, P.P., Cbfbeta interacts with Runx2 and has a critical role in bone development. Genet. 2002, 32, 639–644.
- Miller, J.; Horner, A.; Stacy, T.; Lowrey, C.; Lian, J.B.; Stein, G.; Nuckolls, G.H.; Speck, N.A., The core-binding factor beta subunit is required for bone formation and hematopoietic maturation. Genet. 2002, 32, 645–649.
- Yoshida, C.A.; Furuichi, T.; Fujita, T.; Fukuyama, R.; Kanatani, N.; Kobayashi, S.; Satake, M.; Takada, K.; Komori, T., Core-binding factor beta interacts with Runx2 and is required for skeletal development. Genet. 2002, 32, 633.
- Qin, X.; Jiang, Q.; Matsuo, Y.; Kawane, T.; Komori, H.; Moriishi, T.; Taniuchi, I.; Ito, K.; Kawai, Y.; Rokutanda, S.; Izumi, S.; Komori, T., Cbfb regulates bone development by stabilizing Runx family proteins. Bone Miner. Res. 2015, 30, 706–714.
- Lim, K.E.; Park, N.R.; Che, X.; Han, M.S.; Jeong, J.H.; Kim, S.Y.; Park, C.Y.; Akiyama, H.; Kim, J.E.; Ryoo, H.M.; Stein, J.L.; Lian, J.B.; Stein, G.S.; Choi, J.Y., Core binding factor beta of osteoblasts maintains cortical bone mass via stabilization of Runx2 in mice. Bone Miner. Res. 2015, 30, 715–722.
- Inada, M.; Yasui, T.; Nomura, S.; Miyake, S.; Deguchi, K.; Himeno, M.; Sato, M.; Yamagiwa, H.; Kimura, T.; Yasui, N.; Ochi, T.; Endo, N.; Kitamura, Y.; Kishimoto, T.; Komori, T., Maturational disturbance of chondrocytes in Cbfa1-deficient mice. Dyn. 1999, 214, 279–290.
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; Sato, M.; Okamoto, R.; Kitamura, Y.; Yoshiki, S.; Kishimoto, T., Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764.
- Otto, F.; Thornell, A.P.; Crompton, T.; Denzel, A.; Gilmour, K.C.; Rosewell, I.R.; Stamp, G.W.; Beddington, R.S.; Mundlos, S.; Olsen, B.R.; Selby, P.B.; Owen, M.J., Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997, 89, 765–771.
- Kim, I.S.; Otto, F.; Zabel, B.; Mundlos, S., Regulation of chondrocyte differentiation by Cbfa1. Dev. 1999, 80, 159–170.
- Maruyama, Z.; Yoshida, C.A.; Furuichi, T.; Amizuka, N.; Ito, M.; Fukuyama, R.; Miyazaki, T.; Kitaura, H.; Nakamura, K.; Fujita, T.; Kanatani, N.; Moriishi, T.; Yamana, K.; Liu, W.; Kawaguchi, H.; Komori, T., Runx2 determines bone maturity and turnover rate in postnatal bone development and is involved in bone loss in estrogen deficiency. Dyn. 2007, 236, 1876–1890.
- Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, T., Runx2 regulates cranial suture closure by inducing hedgehog, Fgf, Wnt, and Pthlh signaling pathway gene expression in suture mesenchymal cells. Mol. Genet. 2018, 28, 896–911.
- Simpson, F.; Kerr, M.C.; Wicking, C., Trafficking, development and hedgehog. Dev. 2009, 126, 279–288.
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P., Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086.
- Razzaque, M.S.; Soegiarto, D.W.; Chang, D.; Long, F.; Lanske, B., Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. Pathol. 2005, 207, 453–461.
- Long, F.; Chung, U.I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P., Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318.
- Rodda, S.J.; McMahon, A.P., Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development (Cambridge, England) 2006, 133, 3231–3244.
- Yang, L.; Tsang, K.Y.; Tang, H.C.; Chan, D.; Cheah, K.S., Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Natl. Acad Sci. U S A 2014, 111, 12097–12102.
- Zhou, X.; von der Mark, K.; Henry, S.; Norton, W.; Adams, H.; de Crombrugghe, B., Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet. 2014, 10, e1004820.
- Maes, C.; Kobayashi, T.; Selig, M.K.; Torrekens, S.; Roth, S.I.; Mackem, S.; Carmeliet, G.; Kronenberg, H.M., Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Cell 2010, 19, 329–344.
- Qin X, Jiang Q, Nagano K, Moriishi T, Miyazaki T, Komori H et al. Runx2 is essential for the transdifferentiation of chondrocytes into osteoblasts. PLoS Genet 2020; 16: e1009169.
- Yoshida, C.A.; Yamamoto, H.; Fujita, T.; Furuichi, T.; Ito, K.; Inoue, K.; Yamana, K.; Zanma, A.; Takada, K.; Ito, Y.; Komori, T., Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 2004, 18, 952–963.
- Yoshida, C.A.; Komori, H.; Maruyama, Z.; Miyazaki, T.; Kawasaki, K.; Furuichi, T.; Fukuyama, R.; Mori, M.; Yamana, K.; Nakamura, K.; Liu, W.; Toyosawa, S.; Moriishi, T.; Kawaguchi, H.; Takada, K.; Komori, T., SP7 Inhibits Osteoblast Differentiation at a Late Stage in Mice. PLoS ONE 2012, 7, e32364.
- Nakashima, K.; Zhou, X.; Kunkel, G.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B., The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 2002, 108, 17–29.
- Kawane, T.; Komori, H.; Liu, W.; Moriishi, T.; Miyazaki, T.; Mori, M.; Matsuo, Y.; Takada, Y.; Izumi, S.; Jiang, Q.; Nishimura, R.; Kawai, Y.; Komori, T., Dlx5 and Mef2 regulate a novel Runx2 enhancer for osteoblast-specific expression. Bone Miner. Res. 2014, 29, 1960–1969.
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y., Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Cell 2005, 8, 739–750.
- Hu, H.; Hilton, M.J.; Tu, X.; Yu, K.; Ornitz, D.M.; Long, F., Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development (Cambridge, England) 2005, 132, 49–60.
- Hill, T.P.; Spater, D.; Taketo, M.M.; Birchmeier, W.; Hartmann, C., Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Cell 2005, 8, 727–738.
- Maeno, T.; Moriishi, T.; Yoshida, C.A.; Komori, H.; Kanatani, N.; Izumi, S.; Takaoka, K.; Komori, T., Early onset of Runx2 expression caused craniosynostosis, ectopic bone formation, and limb defects. Bone 2011, 49, 673–682.
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.; Sakane, C.; Matsuo, Y.; Nagai, K.; Maeno, T.; Date, Y.; Nishimura, R.; Komori, T., Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Rep. 2018, 8, 13551.
- Xiao, G.; Jiang, D.; Gopalakrishnan, R.; Franceschi, R.T., Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. Biol. Chem. 2002, 277, 36181–36187.
- Kim, H.J.; Kim, J.H.; Bae, S.C.; Choi, J.Y.; Kim, H.J.; Ryoo, H.M., The protein kinase C pathway plays a central role in the fibroblast growth factor-stimulated expression and transactivation activity of Runx2. Biol. Chem. 2003, 278, 319–326.
- Park, O.J.; Kim, H.J.; Woo, K.M.; Baek, J.H.; Ryoo, H.M., FGF2-activated ERK mitogen-activated protein kinase enhances Runx2 acetylation and stabilization. Biol. Chem. 2010, 285, 3568–3574.
- Ge, C.; Xiao, G.; Jiang, D.; Yang, Q.; Hatch, N.E.; Roca, H.; Franceschi, R.T., Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. Biol. Chem. 2009, 284, 32533–32543.
- Mundlos, S.; Otto, F.; Mundlos, C.; Mulliken, J.B.; Aylsworth, A.S.; Albright, S.; Lindhout, D.; Cole, W.G.; Henn, W.; Knoll, J.H.; Owen, M.J.; Mertelsmann, R.; Zabel, B.U.; Olsen, B.R., Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997, 89, 773–779.
- Lee, B.; Thirunavukkarasu, K.; Zhou, L.; Pastore, L.; Baldini, A.; Hecht, J.; Geoffroy, V.; Ducy, P.; Karsenty, G., Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Genet. 1997, 16, 307–310.
- Mikasa, M.; Rokutanda, S.; Komori, H.; Ito, K.; Tsang, Y.S.; Date, Y.; Yoshida, C.A.; Komori, T., Regulation of Tcf7 by Runx2 in chondrocyte maturation and proliferation. Bone Miner. Metab. 2011, 29, 291–299.
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.; Komm, B.S.; Javed, A.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B., Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. Biol. Chem. 2005, 280, 33132–33140.
- Lee, M.H.; Kim, Y.J.; Yoon, W.J.; Kim, J.I.; Kim, B.G.; Hwang, Y.S.; Wozney, J.M.; Chi, X.Z.; Bae, S.C.; Choi, K.Y.; Cho, J.Y.; Choi, J.Y.; Ryoo, H.M., Dlx5 specifically regulates Runx2 type II expression by binding to homeodomain-response elements in the Runx2 distal promoter. Biol. Chem. 2005, 280, 35579–35587.
- Selvamurugan, N.; Pulumati, M.R.; Tyson, D.R.; Partridge, N.C., Parathyroid hormone regulation of the rat collagenase-3 promoter by protein kinase A-dependent transactivation of core binding factor alpha1. Biol. Chem. 2000, 275, 5037–5042.
- Krishnan, V.; Moore, T.L.; Ma, Y.L.; Helvering, L.M.; Frolik, C.A.; Valasek, K.M.; Ducy, P.; Geiser, A.G., Parathyroid hormone bone anabolic action requires Cbfa1/Runx2-dependent signaling. Endocrinol.2003, 17, 423–435.
- Merciris, D.; Marty, C.; Collet, C.; de Vernejoul, M.C.; Geoffroy, V., Overexpression of the transcriptional factor Runx2 in osteoblasts abolishes the anabolic effect of parathyroid hormone in vivo. J. Pathol. 2007, 170, 1676–1685.
- Aubin, J.; Triffitt, J., Mesenchymal stem ells and osteoblast differentiation. Bilezikian, JP.; Raisz, LG.; Rodan, GA. ed.; Academic Press: Cambridge, MA,USA
- Xiao, Z.; Awad, H.A.; Liu, S.; Mahlios, J.; Zhang, S.; Guilak, F.; Mayo, M.S.; Quarles, L.D., Selective Runx2-II deficiency leads to low-turnover osteopenia in adult mice. Biol. 2005, 283, 345–356.
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G., Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754.
- Sato, M.; Morii, E.; Komori, T.; Kawahata, H.; Sugimoto, M.; Terai, K.; Shimizu, H.; Yasui, T.; Ogihara, H.; Yasui, N.; Ochi, T.; Kitamura, Y.; Ito, Y.; Nomura, S., Transcriptional regulation of osteopontin gene in vivo by PEBP2alphaA/CBFA1 and ETS1 in the skeletal tissues. Oncogene 1998, 17, 1517–1525.
- Harada, H.; Tagashira, S.; Fujiwara, M.; Ogawa, S.; Katsumata, T.; Yamaguchi, A.; Komori, T.; Nakatsuka, M., Cbfa1 isoforms exert functional differences in osteoblast differentiation. Biol. Chem. 1999, 274, 6972–6978.
- Lee, K.S.; Kim, H.J.; Li, Q.L.; Chi, X.Z.; Ueta, C.; Komori, T.; Wozney, J.M.; Kim, E.G.; Choi, J.Y.; Ryoo, H.M.; Bae, S.C., Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Cell Biol. 2000, 20, 8783–8792.
- Banerjee, C.; McCabe, L.R.; Choi, J.Y.; Hiebert, S.W.; Stein, J.L.; Stein, G.S.; Lian, J.B., Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. Cell Biochem. 1997, 66, 1–8.
- Kern, B.; Shen, J.; Starbuck, M.; Karsenty, G., Cbfa1 contributes to the osteoblast-specific expression of type I collagen genes. Biol. Chem. 2001, 276, 7101–7107.
- Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C et al. Increased bone formation in osteocalcin-deficient mice. Nature 1996; 382: 448-452.
- Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C et al. Endocrine regulation of energy metabolism by the skeleton. Cell 2007; 130: 456-469.
- Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE et al. Endocrine regulation of male fertility by the skeleton. Cell 2011; 144: 796-809.
- Mera P, Laue K, Ferron M, Confavreux C, Wei J, Galan-Diez M et al. Osteocalcin Signaling in Myofibers Is Necessary and Sufficient for Optimum Adaptation to Exercise. Cell metabolism 2016; 23: 1078-1092.
- Diegel, C. R., Hann S, Ayturk UM, Hu JCW, Lim KE, Droscha CJ et al. An osteocalcin-deficient mouse strain without endocrine abnormalities. PLoS Genet2020; 16: e1008361.
- Moriishi T, Ozasa R, Ishimoto T, Nakano T, Hasegawa T, Miyazaki T et al. Osteocalcin is necessary for the alignment of apatite crystallites, but not glucose metabolism, testosterone synthesis, or muscle mass. PLoS Genet 2020; 16: e1008586.
- Komori T. Functions of Osteocalcin in Bone, Pancreas, Testis, and Muscle. Int J Mol Sci 2020; 21.
- Lambert LJ, Challa AK, Niu A, Zhou L, Tucholski J, Johnson MS et al. Increased trabecular bone and improved biomechanics in an osteocalcin-null rat model created by CRISPR/Cas9 technology. Disease models & mechanisms 2016; 9: 1169-1179.