CALR mutations are a revolutionary discovery and represent an important hallmark of myeloproliferative neoplasms (MPN), especially essential thrombocythemia and primary myelofibrosis. To date, several CALR mutations were identified, with only frameshift mutations linked to the diseased phenotype. It is of diagnostic and prognostic importance to properly define the type of CALR mutation and subclassify it according to its structural similarities to the classical mutations, a 52-bp deletion (type 1 mutation) and a 5-bp insertion (type 2 mutation), using a statistical approximation algorithm (AGADIR). Today, the knowledge on the pathogenesis of CALR-positive MPN is expanding and several cellular mechanisms have been recognized that finally cause a clonal hematopoietic expansion.
- calreticulin
- chaperone
- calcium
- myeloproliferative neoplasm
- diagnostics
- thrombocythemia
- artificial intelligence
1. Introduction
In 2013, Klampfl et al used whole exome sequencing in six patients with PMF who were JAK2- and MPL-negative and in all of them somatic CALR mutations in exon 9 were confirmed, mutations were either deletions or insertions. Secondly, the 896 patients with different types of MPNs were screened for the presence of insertion or deletion CALR mutations. CALR mutations were observed in patients with ET and PMF [1]. Similar results were obtained by Nangalia et al who analyzed the results of exome sequencing of DNA in 168 patients with MPNs. CALR mutations were identified in 26 patients with either ET or myelofibrosis (MF) and non-mutated JAK2 or MPL [2]. There were two most common variants: CALR NP_004334.1:p.L367fs*46, representing a 52-bp deletion (type 1 mutation); and CALR NP_004334.1:p.K385fs*47, which resulted from a 5-bp insertion (type 2 mutation) [1][2]. CALR mutations were also recognized in patients with other MPN subtypes and similar diseases, although this is mostly an exceptional event. They were identified in a few patients with chronic myelo-monocytic leukemia and atypical chronic myeloid leukemia [2], myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN) [3], unclassified MPN (MPN-U) [4][5] and in rare cases in patients with PV [6]. They were also identified in patients with refractory anemia with ringed sideroblasts and marked thrombocytosis (RARS-t) [7], although this is a rare finding and probably does not occur in patients with strictly WHO-defined RARS-t [8].
Currently, more than 50 CALR mutations in exon 9 have been confirmed. Most commonly these are +1 frameshift mutations, either deletions or insertions leading to a change in the C-terminal domain of the calreticulin protein [9]. It seems that only the mutations leading to the +1 frameshift have a pathogenic potential, other mutations are usually germ line variants of CALR [9].
Today, mutations that are not type 1 or type 2 are classified according to their resemblance with type 1 or type 2 mutations as type 1-like and type 2-like mutations, respectively [10]. Type 1 CALR mutations are more common. In patients with ET type 1 mutation occurs in 55% of patients whereas type 2 mutation occurs in 35% of ET patients. In patients with PMF type 1 mutation is equally more common and occurs in 75% of patients [11]. Classifying mutations as type 1-like and type 2-like mutations carries a prognostic significance and patients with type 1 and type 1-like mutations have a similar predicted survival. Similarly, the prognosis is similar in patients with type 2 and type 2-like mutations. In patients with PMF type 1 and type 1-like mutations have a favorable prognosis compared to type 2 and type 2-like mutations [12].
CALR mutations are an important diagnostic marker in patients with suspected MPN which was recognized by the 2016 revision to the WHO classification of myeloid neoplasms and acute leukemia [13] which included CALR mutations as one of the major criteria for the diagnosis of ET and PMF. In a retrospective study on 524 patients with suspected MPN our research group confirmed the diagnostic significance of CALR mutations in the diagnosis of MPN, however, it seemed that the testing for the presence of CALR mutations should only be performed in patients with clear clinical and/or laboratory suspicion for MPN as in other patients CALR mutations may be atypical with an unknown clinical significance [14]. At about the same time, a large population-based screening study performed on nearly 20,000 Danish citizens by highly sensitive polymerase chain reaction (PCR) method revealed that type 1 and type 2 CALR mutations can indeed be found in patients without confirmed MPN [15]. In fact, in this study, MPN was not confirmed in 30/32 of CALR-positive patients. All CALR mutations detected were either type 1 or type 2 which are known to cause an MPN phenotype. This study suggests that CALR-positive patients are likely to develop MPN even if the disease is not present at the time of CALR mutation detection. Type 1 and type 2 mutations may therefore represent a pre-MPN state with a potential to develop into overt MPN over time [15].
2. The Clinical Value of CALR Mutations
In the current clinical practice, the vast majority of CALR-positive clinical conditions are ET and PMF. The diagnostic approach to patients with ET and PMF is based on the 2016 WHO criteria [13]. From a clinical point of view most ET patients are referred to a hematologist due to accidentally identified elevated platelets in their blood counts. They can present with erythromelalgia, rarely with enlarged spleen and commonly with thrombotic events [16]. The risk for thrombotic complications in patients with ET exceeds 20% [17]. In PMF, patients often present with hepatosplenomegaly, abdominal tenderness in the left upper quadrant, early satiety, fatigue and bone pain [18]. Blood counts in patients with PMF show anemia and variably peripheral blood leukoerythroblastosis [19]. An important hallmark of PMF is extramedullary hematopoiesis which leads to organomegaly and has its origins in the release of bone marrow precursor cells into the circulation as well as overproduction of cytokines that stimulate hematopoiesis and represent a potential therapeutic target [20][21]. Moreover, thrombotic events can be a presenting feature and occur nearly as often as in patients with ET [22].
A bone marrow biopsy is vital for distinguishing between different MPN subtypes and should be performed in all patients. The histologic focus is mainly on megakaryocyte morphology and bone marrow fibrosis involvement. This can help in distinguishing between ET, pre-fibrotic MF and overt MF which all have different prognosis and treatment approach [23]. In all patients with suspected ET or PMF molecular diagnostic tests revealing driver and non-driver mutations need to be performed that can aid in the diagnosis and management of the disease.
Up to this date, driver mutations in the three previously mentioned genes were recognized in ET and PMF patients. In the remaining 10 to 15% of patients with ET or PMF none of these driver mutations are recognized; these patients are referred to as triple negative [24]. The role of driver mutations was elucidated in the recent years. The lower JAK2 allele burden was shown to be associated with poorer survival in PMF [25][26]. CALR-positive patients compared to JAK2-positive patients are younger, have higher platelet counts and are less likely to be anemic, thrombocytopenic, require transfusions or display leukocytosis [27]. They also have a lower risk of progressing to myelofibrosis or acute leukemia and developing thrombotic complications [28][29]. Their leukemia-free and overall survival is superior [27][28]; however, the survival advantage is restricted to type 1/type1-like CALR mutations [30][31]. In patients with ET CALR mutations are associated with younger age, lower hemoglobin level, white blood count, platelet count and erythropoietin level compared to JAK2-positive ET patients. CALR-positive patients with ET have no polycythemic transformation, a similar risk of myelofibrotic transformation and a significantly lower risk of thrombosis compared to JAK2- positive patients [28]. The incidence of splenomegaly between JAK2- and CALR-positive ET patients is similar [32]. These findings represent a foundation for a risk-based therapeutic approach to patients with ET with CALR mutations representing favorable mutations with a lower risk of thrombotic complications and less need of antithrombotic therapy especially in younger patients with no thrombotic history [33].
Additionally, non-driver mutations affect the disease characteristics. With the aid of NGS several novel molecular biomarkers were identified, some of them carrying a prognostic significance [34]. Some examples are mutations in the genes LNK (SH2B3), TET2, DNMT3A, IDH1/2, CBL and ASXL1 and atypical JAK2 and MPL mutations [35][36][37]. Other genes important in the pathogenesis of MPN are DNA methylation genes (TET2, DNMT3A and IDH1) and chromatin structure regulation genes (EZH2, ASXL1) [38]. In a study by Agarwal et al 12% of JAK2-positive patients had additional mutations in the genes TET2, ASXL1 and SF3B1. Additional mutations were also present in up to 10% of CALR-positive patients, in the genes TET2 or ASXL1 [39]. Revolutionary, Grinfeld et al developed a prognostic model based on the sequencing of 69 myeloid genes. This model considered 63 clinical and genetic variables and created a personally tailored clinical prognosis representing a personalized approach to patient prognosis in MPN [40]. Today, routinely, apart from the main three driver mutations (JAK2, CALR, MPL), other mutations in the common myeloid genes can be determined by using the NGS technology which can confirm the accuracy of the diagnosis and provide prognosis [41]. Currently, mutational information on the presence of non-driver mutations is incorporated into a new prognostic model for ET, the Mutation-Enhanced International Prognostic Scoring System (MIPSS). Mutations in the following genes: SRSF2, SF3B1, U2AF1 and TP53 are considered unfavorable [42]. This model is based on a large cohort of ET patients with unfavorable mutations showing a survival disadvantage [43]. The implementation of this model into clinical practice is yet to be elucidated. The prognostication of PMF, on the other hand, went through several stages as the knowledge of the condition increased. The current most often used prognostic system is a dynamic prognostic model (DIPSS) based on age, the hemoglobin level, leukocyte and blast count and the presence of constitutional symptoms [44]. It can be used at any stage of the disease. The implementation of driver and non-driver mutations into prognostic systems in addition to the karyotype, sex-adjusted hemoglobin levels and focus on transplant population led to the development of the most advanced prognostic systems: mutation-enhanced international prognostic scoring system for transplant-age patients (MIPSS70), the karyotype enhanced MIPSS70 (MIPSS70+ version 2.0 (MIPSSv2)) and the genetically-inspired prognostic scoring system (GIPSS) which are relevant in daily patient management [45][46][47].
The algorithmic approach to patients with suspected CALR-positive MPN is presented in Figure 3.
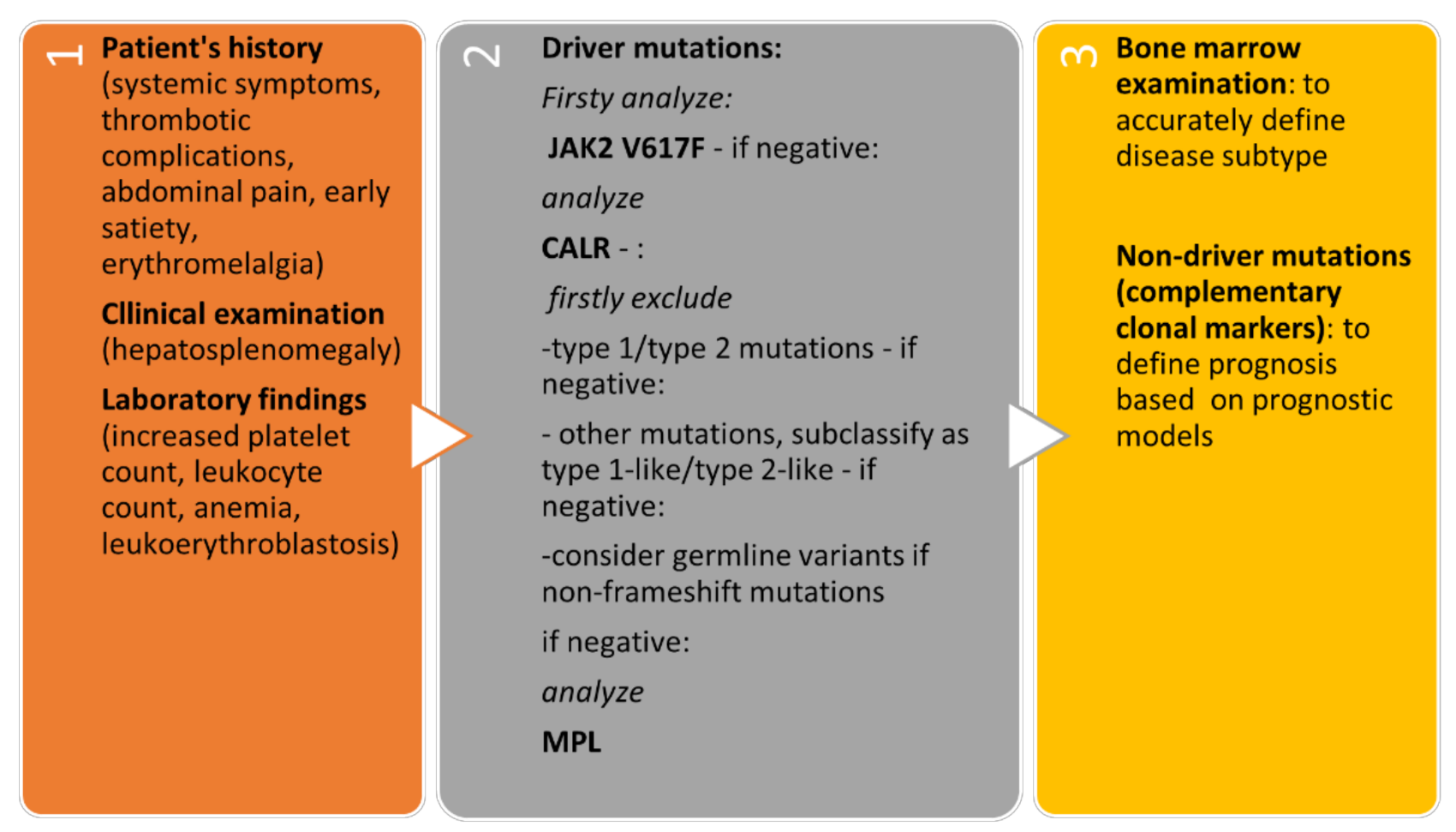
Figure 3. Algorithmic approach to patients with suspected CALR-positive MPN.
3. Conclusions
CALR mutations have a rather short history in hematology; however, due to the rapid development in the knowledge of their role in MPN, it seems their impact is large. The understanding of CALR mutation types led to the development of different assays aiding in their detection. Currently, all the pathological mechanisms causing a diseased phenotype in CALR-positive MPN were not elucidated yet. Nonetheless, it is evident that mutated CALR has many different cellular pathogenic pathways. By binding to MPL and promoting the survival of even misfolded MPL it enables its activation and consequently a constitutive ligand independent activation of JAK2/STAT5 intracellular signaling leading to dysregulated megakaryopoiesis. CALR mutants exhibit their oncogenic potential additionally through homo-multimerization, altered epigenetic regulation and defective calcium storage. As their role in the human cell becomes clearer, novel therapeutic strategies are evolving with potential impact on patients’ outcome. Lastly, in the future the knowledge of the molecular-genetic basis of the disease and their clinical value could become part of the machine learning systems designed to properly diagnose and predict outcome in patients with MPN.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22073371
References
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390.
- Nangalia, J.; Massie, C.; Baxter, E.; Nice, F.; Gundem, G.; Wedge, D.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.; et al. SomaticCALRMutations in Myeloproliferative Neoplasms with NonmutatedJAK2. N. Engl. J. Med. 2013, 369, 2391–2405.
- Heuser, M.; Panagiota, V.; Koenecke, C.; Fehse, B.; Alchalby, H.; Badbaran, A.; Shahswar, R.; Stadler, M.; Eder, M.; Göhring, G.; et al. Low frequency of calreticulin mutations in MDS patients. Leukemia 2014, 28, 1933–1934.
- Kim, S.Y.; Im, K.; Park, S.N.; Kwon, J.; Kim, J.-A.; Lee, D.S. CALR, JAK2, and MPL Mutation Profiles in Patients with Four Different Subtypes of Myeloproliferative Neoplasms. Am. J. Clin. Pathol. 2015, 143, 635–644.
- Lin, Y.; Liu, E.; Sun, Q.; Ma, J.; Li, Q.; Cao, Z.; Wang, J.; Jia, Y.; Zhang, H.; Song, Z.; et al. The Prevalence ofJAK2, MPL, and CALR Mutations in Chinese Patients WithBCR-ABL1–Negative Myeloproliferative Neoplasms. Am. J. Clin. Pathol. 2015, 144, 165–171.
- Broséus, J.; Park, J.-H.; Carillo, S.; Hermouet, S.; Girodon, F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood 2014, 124, 3964–3966.
- Broséus, J.; Lippert, E.; Harutyunyan, A.S.; Jeromin, S.; Zipperer, E.; Florensa, L.; Milosevic, J.D.; Haferlach, T.; Germing, U.; Luño, E.; et al. Low rate of calreticulin mutations in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia 2014, 28, 1374–1376.
- Patnaik, M.M.; Belachew, A.; Finke, C.; Lasho, T.L.; Hanson, A.C.; Tefferi, A. CALR mutations are infrequent in WHO-defined refractory anemia with ring sideroblasts. Leukemia 2014, 28, 1370–1371.
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679.
- Pietra, D.; Rumi, E.; Ferretti, V.; Di Buduo, A.C.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438.
- Cabagnols, X.; Defour, J.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.; et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2015, 29, 249–252.
- Guglielmelli, P.; Rotunno, G.; Fanelli, T.; Pacilli, A.; Brogi, G.; Calabresi, L.; Pancrazzi, A.; Vannucchi, A.M. Validation of the differential prognostic impact of type 1/type 1-like versus type 2/type 2-like CALR mutations in myelofibrosis. Blood Cancer J. 2015, 5, 360.
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405.
- Mikic, T.B.; Pajic, T.; Sever, M. CALR mutations in a cohort of JAK2 V617F negative patients with suspected myeloproliferative neoplasms. Sci. Rep. 2019, 9, 1–9.
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479.
- Michiels, J.J.; Van Genderen, P.J.J.; Lindemans, J.; Van Vliet, H.H.D.M. Erythromelalgic, Thrombotic and Hemorrhagic Manifestations in 50 Cases of Thrombocythemia. Leuk. Lymphoma 1996, 22, 47–56.
- Tefferi, A.; Elliott, M. Thrombosis in Myeloproliferative Disorders: Prevalence, Prognostic Factors, and the Role of Leukocytes and JAK2V617F. Semin. Thromb. Hemost. 2007, 33, 313–320.
- Tefferi, A. Myelofibrosis with Myeloid Metaplasia. N. Engl. J. Med. 2000, 342, 1255–1265.
- Kvasnicka, H.M.; Thiele, J. Prodromal myeloproliferative neoplasms: The 2008 WHO classification. Am. J. Hematol. 2009, 85, 62–69.
- Tefferi, A. Pathogenesis of Myelofibrosis with Myeloid Metaplasia. J. Clin. Oncol. 2005, 23, 8520–8530.
- Jutzi, J.S.; Mullally, A. Remodeling the Bone Marrow Microenvironment—A Proposal for Targeting Pro-inflammatory Contributors in MPN. Front. Immunol. 2020, 11.
- Kc, D.; Falchi, L.; Verstovsek, S. The underappreciated risk of thrombosis and bleeding in patients with myelofibrosis: A review. Ann. Hematol. 2017, 96, 1595–1604.
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Ruggeri, M.; Rodeghiero, F.; D’Amore, E.S.; Randi, M.L.; Bertozzi, I.; et al. Survival and Disease Progression in Essential Thrombocythemia Are Significantly Influenced by Accurate Morphologic Diagnosis: An International Study. J. Clin. Oncol. 2011, 29, 3179–3184.
- Langabeer, S.E. Chasing down the triple-negative myeloproliferative neoplasms: Implications for molecular diagnostics. Jak-stat 2016, 5, e1248011.
- Rozovski, U.; Verstovsek, S.; Manshouri, T.; Dembitz, V.; Bozinovic, K.; Newberry, K.; Zhang, Y.; Bove, J.E.; Pierce, S.; Kantarjian, H.; et al. An accurate, simple prognostic model consisting of age, JAK2, CALR, and MPL mutation status for patients with primary myelofibrosis. Haematology 2016, 102, 79–84.
- Guglielmelli, P.; Barosi, G.; Specchia, G.; Rambaldi, A.; Coco, F.L.; Antonioli, E.; Pieri, L.; Pancrazzi, A.; Ponziani, V.; Delaini, F.; et al. Identification of patients with poorer survival in primary myelofibrosis based on the burden of JAK2V617F mutated allele. Blood 2009, 114, 1477–1483.
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Knudson, A.R.; Ketterling, R.; Hanson, C.H.; Maffioli, M.; Caramazza, D.; Passamonti, F.; Pardanani, A. CALR vs JAK2 vs MPL-mutated or triple-negative myelofibrosis: Clinical, cytogenetic and molecular comparisons. Leukemia 2014, 28, 1472–1477.
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551.
- Finazzi, M.C.; Carobbio, A.; Cervantes, F.; Isola, I.M.; Vannucchi, A.M.; Guglielmelli, P.; Rambaldi, A.; Finazzi, G.; Barosi, G.; Barbui, T. CALR mutation, MPL mutation and triple negativity identify patients with the lowest vascular risk in primary myelofibrosis. Leukemia 2014, 29, 1209–1210.
- Tefferi, A.; Lasho, T.L.; Tischer, A.; Wassie, E.A.; Finke, C.M.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.D. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood 2014, 124, 2465–2466.
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Szuber, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1,095 patients. Am. J. Hematol. 2018, 93, 348–355.
- Saki, N.; Shirzad, R.; Rahim, F.; Malehi, A.S. Estimation of diagnosis and prognosis in ET by assessment of CALR and JAK2V617F mutations and laboratory findings: A meta-analysis. Clin. Transl. Oncol. 2017, 19, 874–883.
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2020, 95, 1599–1613.
- Palumbo, G.A.; Stella, S.; Pennisi, M.S.; Pirosa, C.; Fermo, E.; Fabris, S.; Cattaneo, D.; Iurlo, A. The Role of New Technologies in Myeloproliferative Neoplasms. Front. Oncol. 2019, 9, 321.
- Pardanani, A.; Lasho, T.L.; Finke, C.M.; Oh, S.T.; Gotlib, J.; Tefferi, A. LNK mutation studies in blast-phase myeloproliferative neoplasms, and in chronic-phase disease with TET2, IDH, JAK2 or MPL mutations. Leukemia 2010, 24, 1713–1718.
- Tefferi, A.; Pardanani, A. Myeloproliferative Neoplasms. JAMA Oncol. 2015, 1, 97–105.
- Chang, Y.-C.; Lin, H.-C.; Chiang, Y.-H.; Chen, C.G.-S.; Huang, L.; Wang, W.-T.; Cheng, C.-C.; Lin, J.; Chang, Y.-F.; Chang, M.-C.; et al. Targeted next-generation sequencing identified novel mutations in triple-negative myeloproliferative neoplasms. Med. Oncol. 2017, 34, 83.
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228.
- Agarwal, R.; Blombery, P.; McBean, M.; Jones, K.; Fellowes, A.; Doig, K.; Forsyth, C.; Westerman, D.A. Clinicopathological differences exist between CALR- and JAK2-mutated myeloproliferative neoplasms despite a similar molecular landscape: Data from targeted next-generation sequencing in the diagnostic laboratory. Ann. Hematol. 2017, 96, 725–732.
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430.
- Gardner, J.-A.; Peterson, J.D.; Turner, S.A.; Soares, B.L.; Lancor, C.R.; Dos Santos, L.L.; Kaur, P.; Ornstein, D.L.; Tsongalis, G.J.; De Abreu, F.B. Detection ofCALRMutation in Clonal and Nonclonal Hematologic Diseases Using Fragment Analysis and Next-Generation Sequencing. Am. J. Clin. Pathol. 2016, 146, 448–455.
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Coltro, G.; Finke, C.M.; Loscocco, G.G.; Sordi, B.; Szuber, N.; Rotunno, G.; Pacilli, A.; et al. Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br. J. Haematol. 2020, 189, 291–302.
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30.
- Cervantes, F.; Dupriez, B.; Pereira, A.; Passamonti, F.; Reilly, J.T.; Morra, E.; Vannucchi, A.M.; Mesa, R.A.; Demory, J.-L.; Barosi, G.; et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009, 113, 2895–2901.
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642.
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770.
- Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Mudireddy, M.; Mannarelli, C.; Nicolosi, M.; Pacilli, A.; Pardanani, A.; Rumi, E.; Rosti, V.; et al. MIPSS70: Mutation-Enhanced International Prognostic Score System for Transplantation-Age Patients with Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 310–318.