Immune checkpoint inhibitors have revolutionized the treatment landscape for many solid tumors. Amongst gastric cancer subtypes, tumors with microsatellite instability and Epstein Barr Virus positive tumors provide the strongest rationale for responding to immunotherapy. Various predictive biomarkers such as mismatch repair status, programmed death ligand 1 expression, tumor mutational burden, assessment of tumor infiltrating lymphocytes and circulating biomarkers have been evaluated. However, results have been inconsistent due to different methodologies and thresholds used. Clinical implementation therefore remains a challenge. The role of immune checkpoint inhibitors in gastric cancer is emerging with data from monotherapy in the heavily pre-treated population already available and studies in earlier disease settings with different combinatorial approaches in progress. Immune checkpoint inhibitor combinations with chemotherapy (CT), anti-angiogenics, tyrosine kinase inhibitors, anti-Her2 directed therapy, poly (ADP-ribose) polymerase inhibitors or dual checkpoint inhibitor strategies are being explored.
- immune checkpoint inhibitors
- gastric cancer
- Epstein Barr Virus
- tumor mutational burden
- microsatellite instability
- predictive biomarkers
- CAR T cell therapy
- vaccines
1. Introduction
Overview of Gastric Cancer Classification and Relevance for Immunotherapy
Gastric cancer (GC) is a leading global cause of morbidity and mortality [1]. In 2020, over a million people were diagnosed with GC (representing almost 6% of all cancer diagnoses), and nearly 800,000 patients died due to this disease (representing 8.2% of all cancer deaths) [2]. Worldwide, GC is particularly prevalent in East Asia and central/Eastern Europe.
The Lauren classification, published in 1965, differentiates gastric adenocarcinoma into two distinct types, termed the intestinal and diffuse subtypes [3]. The intestinal type is most common, present in over half of the patients and characterized by microscopic glandular structures, with infiltrating capacity of the mesenchymal tissues [4]. The diffuse subtype accounts for a third of cases and is characterized by poor differentiation and poorly cohesive malignant cells with invasive capacity [5]. In general, the intestinal type is associated with exogenous risk factors such as Helicobacter pylori, while the diffuse subtype encompasses a hereditary familial pattern related to germline pathogenetic mutations of the E-cadherin (CDH1) and αE-catenin (CTNNA1) genes [6]. While these subtypes of GC are associated with different carcinogenesis mechanisms and disease biology, this classification, along with the subsequent World Health Organization classification of GC, has not translated into distinct subtype-driven treatment strategies [7,8]. More recently, following comprehensive molecular profiling, The Cancer Genome Atlas (TCGA) defined four distinct subtypes of gastric cancer: Epstein-Barr virus (EBV) positive, microsatellite unstable tumors (MSI), genomically stable tumors (GS) and tumors with chromosomal instability (CIN) [9]. Significant overlap was seen between the histologically determined Lauren’s diffuse variant and the molecular GS TCGA subtype [10]. Interestingly, certain molecular subtypes were most commonly detected in specific anatomic locations with EBV positive tumors more likely to be in the gastric fundus or body and CIN tumors in the cardia [9]. Although the molecular classification of gastric cancer has not directly changed clinical practice, it has provided an important platform to identify novel molecular targets and pave the way for innovative clinical trial design with the incorporation of biomarker enrichment stratification strategies. EBV-positive and MSI tumors are associated with signatures suggestive of an immune responsive profile [11]. A hyper-mutated DNA phenotype is defined as 20.5 mutations/Mb in GC and is a phenotype typical of most MSI tumors [12]. The MSI high (MSI-H) phenotype is most commonly related to epigenetic silencing of the mismatch repair gene, MLH1, rather than germline mutation (i.e., Lynch syndrome) [13]. The presence of a higher number of somatic mutations has been associated with a better prognosis [14] and an increased susceptibility to immune-activating antineoplastic treatments [15]. Currently, patients with MSI gastric cancer can benefit from established immunotherapy approaches with anti-programmed death-1 (anti-PD-1) immune-checkpoint inhibitors [16]. Rather than a hypermutated phenotype, EBV-positive tumors (accounting for 9% of GC) have a profile favoring immunotherapy in view of their high expression of membrane immune-checkpoint molecules such as programmed death ligand-1 (PD-L1) and 2. Key molecular features of EBV-positive tumors include the expression of virus-associated antigens (e.g., nuclear antigen 1, latent membrane protein 2A), the extensive methylation of viral and host genome and the epigenetic regulation of specific cytosine-phosphatidyl-guanosine (CpG) DNA islands through methylation mechanisms [17]. The pattern of DNA methylation of CpG has been associated with anti-tumor immune-activation, with predictive and prognostic significance [18,19]. Therefore, MSI and EBV-positive tumors have been proposed as chief candidates for immunotherapy trials, though not exclusively, for their intrinsic immune-mediated biology [11]. The advent of immunotherapy in oncology has in fact been embraced in most if not all tumor types and disease settings [20]. The identification of an immune-signature or predictive factors of immune-response in patients with GC have been identified as a research priority given that it is a tumor type associated with poor prognosis when diagnosed at an advanced stage and any benefit derived from chemotherapy (CT) is very limited [21]. While advancements in the development of pharmacotherapies have improved overall survival (OS) and quality of life, the low proportion of patients alive after two years from the diagnosis of metastatic disease remains a cause for concern [22,23].
The strategies implemented to enhance the immune response against tumors, including GC, aim to re-orient the immune-system response, by dampening the suppressive regulatory molecules and enhancing a stimulating milieu [24]. This strategy has been pursued by developing a number of immune-checkpoint inhibitors [e.g., PD-1, cytotoxic T-lymphocyte antigen-4 (CTLA-4)], a class of molecules capable of acting on several immune cells and (re-)activating an effective antineoplastic response [25]. This strategy is particularly beneficial in tumors exerting immune-activating signatures and/or recognized by the immune-system as foreign, and therefore regulated by the immune-response [26].
Another therapeutic approach is based on the bioengineering of immune-competent cells against specific tumor- associated antigens [27]. The principal expression of this approach is represented by the Chimeric Antigen Receptor T-cells (CAR-T) constructs. CAR-T are genetically engineered T-cells designed to direct the specific immune-response against tumor- antigens, thereby inducing an artificial acquired antineoplastic immune response, through cytotoxic activity. Though still widely experimental in solid neoplasms, the clinical implementation of CAR-T cells for hematological malignancies has paved a new way of cancer immunotherapy, due to the durable responses seen in some cases, the different patterns of response observed [28] as well as the specific safety profile which needs to be considered and the structural efforts required to build and deliver cell-based treatments [29].
2. Biomarkers of Response to Immunotherapy in Gastric Cancer
The characterization of immune-related biomarkers is becoming increasingly important in the multi-modality treatment of advanced GC (Figure 1) [30].
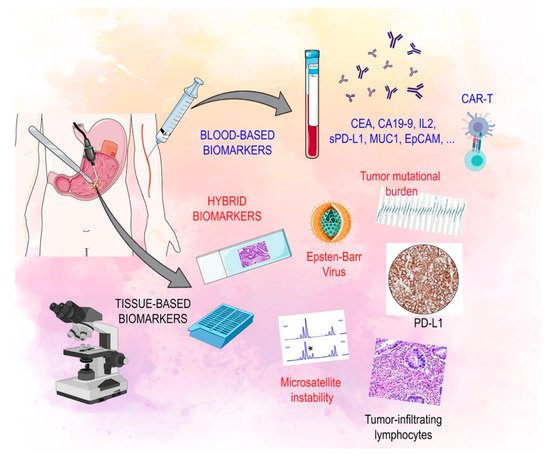
2.1. Tissue Based Biomarkers
2.2. Circulating Biomarkers
Circulating molecules and their role in predicting response to immunotherapy is a topic of great interest and is an area that still has not been studied in depth. These soluble factors can be released from both tumor cells and immune cells and may provide a simple method to evaluate the dynamic behavior of the immune system in cancer patients during treatment and avoid the need for invasive procedures [66]. Much effort is being spent on identifying primary responders to immunotherapy at a relatively early treatment timepoint. Circulating biomarkers, some of which are currently used in clinical practice, such as pepsinogen, carcinoembryonic antigen (CEA), carbohydrate antigen 19-9 (CA19-9) and soluble IL-2, are not accurate enough to predict prognosis in GC [67,68,69].
Recent evidence has shown that patients with GC have higher serum soluble PD-L1 (sPD-L1) concentrations than healthy controls. Moreover, both elevated tissue PD-L1 and serum sPD-L1 were independent prognostic factors for poor OS and poor DFS in GC patients who underwent surgery [70,71,72]. Lymphocyte activation gene 3 (LAG3) is a checkpoint receptor localized on activated T cell surfaces and NK cells. The soluble variant, in turn, can have a regulatory function on immune cells [73,74]. Its role has been investigated in GC patients. High sLAG-3 expression is associated with a better prognosis in GC and its expression was positively correlated with IL-12 and IFN-γ production in GC patients. In a recent in vivo experiment sLAG3 was shown to be able to promote the activation of CD8 T cells and the production of INF and IL-12, resulting in tumor growth inhibition [75].
While the prognostic value of soluble checkpoints is under investigation in several solid tumors, the question that remains to be answered is whether soluble checkpoints can predict response to treatment. Given that the immune system is a key factor involved in the response to treatments such as immunotherapy and CT, there is a clear rationale to suggest that such soluble markers could be biomarkers of response to treatment [76]. A study including 11 patients with NSCLC and 9 patients with GC treated with an anti-PD1 agent showed that pre-treatment levels of sPD-L1 were not associated with OS in these patients. However, reduction in plasma sPD-L1 levels was significantly associated with tumor response after four cycles of treatment [77]. A study including 68 patients with metastatic GC eligible for first line CT analyzed baseline level of sPDL1 and the dynamic changes during therapy. Patients with low levels of sPD-L1 at diagnosis showed a better OS and PFS than patients with a high sPDL1. Patients whose sPDL1 increased after the first cycle of CT showed worse PFS and OS. This result suggests that soluble checkpoints may be the ideal method of studying the immune system as an extremely dynamic entity allowing real-time, non-invasive monitoring during cancer treatment [78]. Takahashi et al. confirmed in their study that high serum levels of sPD-L1 correlated with worse OS in patients with metastatic GC treated with first-line CT [79]. These data suggest the possibility of individualizing the therapeutic choice based on the immunological profile, thereby leading to promising new combination strategies in the near future.
Immunotherapeutics in solid tumors is constantly evolving due to the introduction of new technologies to manipulate the patient’s immune system to attack cancer cells. Tumor antigen vaccines are currently being studied in several solid tumors. They are created from cancer cells’ pure tumor antigens [80]. The antitumor activity of tumor peptide vaccines, such as G17DT, vascular endothelial growth factor receptor (VEGFR) and OTSGC-A24, have been investigated in GC patients. G17DT is a vaccine able to promote an immune response against gastrin, a hormone involved in carcinogenesis and progression in GC [81,82,83]. A phase II/III study (NCT00042510) reported that G17DT is able to induce efficient anti-gastrin antibody production and is able to inhibit tumor proliferation and progression [84]. A multi-center study showed that the combination of G17DT and platinum-5FU CT prolonged the median time-to-progression and median survival time for patients with unresectable cancer of the stomach or gastroesophageal junction, compared to platinum-5FU CT alone. Therefore, the FDA approved the fast track designation for the vaccine G17DT in February 2003 [85]. Another peptide vaccine involving the use of VEGFR 1 and 2, receptors of the VEGF angiogenic factor, has been investigated. In a phase I/II study, the administration of the VEGFR1/2 peptide vaccine in combination with CT induced a cytotoxic T cell response. In the 82% of patients with a cytotoxic T lymphocyte response to VEGFR2-169 peptide, time to progression and OS were significantly prolonged compared to those without such a response [86]. Such findings are encouraging, although it should be noted that only 22 patients were included. A phase I/Ib study (NCT01227772) evaluated OTSGC-A24, which is thought to be able to target several specific tumor antigens, such as forkhead box M1, DEP domain containing 1, kinesin family member 20A, URLC10 and VEGFR1. Although the treatment was well tolerated, no radiological responses were observed [83].
An innovative immunotherapeutic strategy uses adoptive T cell therapy to overcome the immune-evasion mechanisms mediated by cancer cells. T lymphocytes are removed from patients and modified in vitro in order to activate specific immune cells. Then, the modified activated T cells are administered to patients, thereby eliciting a tumor response against cancer [87]. Chimeric antigen receptor-T (CAR-T) cell therapy was shown to be effective in hematologic disease and it is actually under investigation in several solid tumors [88,89].
In GC, several antigens, including human epidermal growth factor receptor 2 (HER2), carcinoembryonic antigen (CEA), mucin 1 (MUC1) and epithelial cell adhesion molecule (EpCAM), have been used as targets for CAR-T. The anti-HER2 CAR-modified T cell was evaluated in many pre-clinical studies [90]. Clinical studies are now evaluating it in GC patients (NCT02713984, NCT01935843, NCT00889954). CEA-specific CAR-T cells were confirmed to be active in pre-clinical studies in mice with GC. Since then, a clinical trial is ongoing (NCT02349724) to define the correct dose and safety profile [91,92]. MUC1 and EpCAM are transmembrane glycoproteins expressed in different solid tumors, but in GC they are markers of aggressive disease. Clinical Phase I/II trials (NCT02617134, NCT02725125) are evaluating EpCAM and MUC1 modified CAR-T in solid tumors expressing these targets [93].
This entry is adapted from the peer-reviewed paper 10.3390/jcm10071412