2.1. Conventional T Cells
Adoptive transfer of ex vivo expanded T cells from the natural repertoire is a promising approach to treat a variety of cancers. However, this requires stimulatory signals to promote in vitro proliferation of T cells that impart a T-cell differentiation program that is susceptible to inducing T-cell dysfunction. Hence, understanding how T-cell dysfunction develops or is programmed by the ex vivo expansion process is imperative. Strategies to promote optimal differentiation and functionality are susceptible to promoting better function and persistence of the transferred cells.
Allogenic hematopoietic cell transplantation is not only the first established form of ACT, it has also served as an ideal setting to advance the field. One of the greatest successes of ACT in the wake of allogeneic hematopoietic cell transplantation is the treatment of immunosuppression-associated opportunistic virus reactivations. Antiviral T cells expanded ex vivo have proven to be highly effective in controlling such viral reactivations in both hematopoietic and organ transplantation [
68,
69,
70,
71,
72]. Over the years, manufacture duration for these virus-specific T-cell products has gone from three months to around 10 days [
73]. It is also possible to directly enrich virus-specific T cells from a healthy donor apheresis product and administer the T cells without further expansion [
74]. Most of the published experience with virus-specific T cells reports on the generation of cellular products manufactured from a robust memory repertoire in healthy donors. Such memory cells readily expand in the culture and show limited evidence of T-cell dysfunction at the time of infusion. In hematopoietic cell transplantation, long-term persistence of the transferred cells, thereby establishing long-lasting memory in the recipients, has been shown when the virus-specific T cells are prepared from the original stem cell donor [
69]. However, the stimulation and expansion of T cells from naïve repertoire has proven to be more difficult. Although clearly feasible for virus-specific T cells [
75,
76], the generation and expansion of T cells targeting tumor-associated antigens (TAA) or transplantation antigens from naïve repertoires require more elaborate culture processes [
1,
2,
77,
78,
79].
Alloreactive donor T cells can recognize major histocompatibility complex (MHC)-bound polymorphic peptides derived from the host proteome, known as minor histocompatibility antigens (MiHAs) [
80,
81,
82,
83]. Most of the molecularly characterized MiHAs are encoded by autosomal genes that differ between patient and donor secondary to germline-encoded non-synonymous single nucleotide polymorphisms (ns-SNP). Although potent immunogenic antigens, up to five rounds of ex vivo weekly stimulation with antigen-loaded dendritic cells and the presence of interleukin (IL)-2 are often needed to generate high numbers of specific CD8
+ T cells [
2]. However, despite the immunogenic nature of these antigens, protocols often generate dysfunctional cells that highly impede cell functionality and persistence after adoptive transfer [
78,
79]. Repeated ex vivo antigen exposure in the context of stimulatory cytokines eventually blunts T-cell growth and leads to terminal differentiation and exhaustion marker expression, especially in antigen-specific T cells in the culture [
2].
It is now generally accepted that optimal therapeutic effects are achieved when the ex vivo expanded T cells maintain features associated with early memory differentiation (Tscm or Tcm) [
3,
84]. The clinical outcome might thus depend more on the antigen-specific T-cell early differentiation phenotype, leading to a better ability to proliferate and persist in vivo, rather than on the bulk number of infused cells [
85]. As such, careful design of ex vivo culture conditions may promote the acquisition of a favorable differentiation status. Exogenous cytokines, small molecules altering cell signaling, metabolic modulation, and epigenetic modification during the ex vivo priming and expansion phase may confer early memory features [
86]. We and others have demonstrated that exogenous exposure to IL-21 can limit terminal differentiation of antigen-specific T cells, which can increase their in vivo persistence and promote phenotypic and functional characteristics associated with long-lived memory T cells [
77,
87]. The histone deacetylase inhibitor (HDACi) combined with IL-21 can also reprogram differentiated CD8
+ T cells into central memory-like T cells. This is achieved through an increase in histone H3 acetylation and chromatin accessibility at the
CD28 promoter region. It is then followed by an IL-21-mediated phosphorylation of the signal transducer and activator of transcription 3 (STAT3) binding to the
CD28 region, and a resulting memory-associated transcriptional signature [
88]. As the epigenetic landscape of the terminal effector and exhausted T cells is being defined, it is likely that several interventions involving epigenetic modulation are introduced in T-cell manufacturing protocols [
89,
90]. As such, the inhibition of DNA methylation programs responsible for exhaustion-associated signaling can be a strategy to improve T-cell function [
91]. A dominant feature of memory differentiation promoting interventions is the mitigation of activation signals. A brief exposure to the immunosuppressive cytokine transforming factor beta (TGFβ) promotes the Tcm differentiation of ex vivo expanded human T cells [
86]. Along the same lines, AKT inhibition has been shown to increase early T-cell memory features [
92]. The association of AKT inhibition and early memory T-cell differentiation also highlights how the modulation of pathways associated with cell growth and metabolism are attractive targets for programming T-cell fates. Likewise, limiting glycolysis at the expense of oxidative phosphorylation during ex vivo T-cell expansion has been suggested to improve the fitness of adoptively transferred T cells (reviewed in [
26]), and limiting reactive oxygen species metabolism with N-acetylcysteine during T-cell manufacturing can favor early memory T-cell formation [
93].
Nonetheless, relying solely on phenotyping may be misleading. The proportion of effector memory or central memory phenotype cells does not necessarily correlate with ex vivo loss of antigen-specific cells or a decline in their functionality [
2]. Although repeated antigenic stimulation of MiHA-specific T cells may lead to terminal differentiation, prolonging the expansion phase in the absence of antigenic stimulation can decrease T-cell proliferation despite limited expression of inhibitory receptors and the preservation of polyfunctional cytokine secretion by the remaining antigen-reactive cells. Thus, the analysis of both phenotypic and functional properties of T cells prior to ACT may best inform about the potency of the T-cell product [
85]. In addition, it has been shown that a fraction of terminally differentiated melanoma-specific or leukemia-specific CTL clones after ex vivo expansion appears to revert back to a central memory type in vivo after ACT, potentially conferring clinical benefits [
77,
94].
As an alternative to limiting the development of dysfunction, some groups have concentrated on cell reprogramming. A terminally differentiated or exhausted T cell may be induced into a pluripotent stem cell (iPSC), which enables re-differentiation into a naïve or central-memory phenotype T cell with the re-expression of CCR7, CD27, and CD28 and no exhaustion markers [
95,
96,
97,
98]. An interesting characteristic of iPCSs generated from lymphocytes is their ability to keep the rearranged TCR loci of the parental cells, which remain unchanged during in vitro differentiation [
95,
97]. As such, antigen-specific CTL clones can be preselected and reprogrammed into iPSC (T-iPSCs) with the Yamanaka transcription factors (Oct3/4, SOX2, KLF4, and c-MYC) [
95,
96,
97,
99,
100]. These new CTLs then have longer telomeres than the original cells, with higher proliferative and functional capacities [
95]. Furthermore, exhausted T cells turned into T-iPSCs are functionally able to respond to antigen-specific stimulation [
101]. However, a caveat with the use of iPSC in cell therapy is the risk of only partial re-differentiation and teratoma formation post-transfer (A).
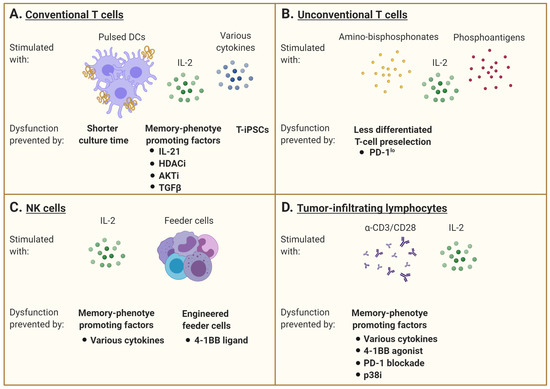
Figure 2. Interventions aiming at limiting T-cell dysfunction for adoptive immunotherapy. (
A) Conventional αβ T cells are generally stimulated with specific peptides that may be presented by professional antigen-presenting cells such as dendritic cells (DCs) and supported by IL-2 supplementation and/or other cytokines. However, repetitive or chronic antigen stimulation promotes the gradual accumulation of dysfunctional cells over time. Many interventions are under investigation to maintain the cells in a less differentiated state. A shorter culture time allows for the maintenance of a higher percentage of healthy cells, but this may limit the number of cells manufactured. The addition of various factors to promote a memory phenotype differentiation is also evaluated, including the use of IL-21, TGFβ, histone deacetylase (HDAC) inhibitors, or AKT inhibitors. The rejuvenation of dysfunctional T cells by induced pluripotent stem cell technology can also generate less differentiated cells. (
B) Unconventional γδ T cells are stimulated with amino-bisphosphonates and phosphoantigens, combined with IL-2. The pre-selection of T cells with a favorable phenotype, excluding cells expressing inhibitory receptors such as PD-1, has been investigated to limit the development of dysfunctional features. (
C) Natural killer (NK) cells are mainly activated with IL-2 and feeder cells. Supplementation with various cytokines or agonists of co-stimulatory molecules (e.g., 4-1BB) can shape their differentiation status and modulate their receptor expression pattern. The engineering of feeder cells as well as the NK cells themselves to express these factors is also underway. (
D) Tumor-infiltrating lymphocytes are stimulated with anti-CD3 with or without anti-CD28, with IL-2 supplementation. As for conventional T cells, the addition of various cytokines can promote a memory phenotype differentiation. The combination of a 4-1BB agonist and PD-1 blockade can also prevent the development of dysfunction. Inhibition of p38MAPK signaling has further shown to limit development of senescence features. This figure was created with
BioRender.com.
2.2. Unconventional T Cells
γδ TCR-expressing T lymphocytes is another T-cell subtype with effector and regulatory functions and the ability to infiltrate tumors. Adoptive cell therapy with human γδ T cells expressing a Vγ2Vδ2 TCR has shown promise because of their capacity to recognize and kill most types of tumors in an MHC-unrestricted manner [
102]. Vγ9Vδ2 T cells are relatively abundant in human blood and can be easily ex vivo expanded in response to amino-bisphosphonates (N-BPs) or phosphoantigens (PAgs). However, culture conditions, timing, and dosage of N-BPs or PAgs, as well as added co-stimulators such as IL-2, may result in different phenotypes and effector cell characteristics [
103,
104]. Results of in vitro expansion are often highly donor dependent and may also predict the respective in vivo expansion efficacy, which can be additionally restricted in cancer patients. Currently, optimal doses of N-BPs or PAgs as well as IL-2 have not yet been determined for ex vivo expansion. The efficacy of stimulation may depend on drug concentration as well as duration of exposure, which have to be individualized [
105]. The role of additional systemic application of N-BPs in the context of adoptive cell transfer strategies also remains elusive. On one side, it has been reported to promote the engraftment of ex vivo-stimulated and adoptively transferred human cells in mice, but on the other side, there are indications that repetitive application of these drugs in vivo induces Vγ9Vδ2 T-cell exhaustion [
103]. As an alternative, some groups tried to adoptively transfer PD-1
lo Vδ2
+ T cells to bypass the tumor immunosupressive environment in vivo (B) [
106].
Other T cells with “innate-like” characteristics can recognize vitamin metabolites, small phosphoantigens, and lipid antigens presented within various highly conserved and non-polymorphic MHC class I-like molecules [
107,
108,
109,
110]. One of the best characterized subsets is invariant natural killer T (iNKT) cells, which recognize lipid antigens bound within the antigen-presenting molecule CD1d [
111]. These cells utilize the near-germline TCRα rearrangement Vα24-Jα18 combined with a limited TCRβ repertoire and are functionally defined by their ability to respond to galactosylceramide (α-GalCer) when presented by CD1d molecules [
107,
108,
109,
110,
112,
113]. They are potent cytokine secretors that bridge innate and adaptive immunity [
109,
111]. These cells have thus been investigated as cell transfer therapy products [
114,
115,
116,
117,
118,
119]. However, whether these cells develop dysfunctional features prior, during, and after therapy is still unclear. Nevertheless, since iNKT cells have been shown to limit GVHD [
120], there is a growing interest to use them as a platform for cell engineering [
121].
The development of ACT using other lymphoid cells is also rapidly expanding. Innate lymphoid cells (ILCs) derive from common lymphoid progenitors in the bone marrow that lack other lineage markers and genetically rearranged antigen receptors. They are defined according to their cytokine production pattern as well as unique transcription factors [
122,
123,
124]. Group 1 ILCs (ILC1s) secrete IFNγ and express the transcription factor T-bet, whereas ILC2s produce IL-5 and IL-13 and require expression of Gata3 [
125,
126,
127,
128]. ILC3s generate IL-22 and IL-17 and are defined by the expression of RORγt, as with lymphoid tissue-inducer cells, which also express IL-7Rα [
129,
130,
131]. Since ILCs can respond to many danger signals such as innate immune cells and secrete cytokines such as T cells despite the lack of TCR, different therapeutic approaches aim at targeting these cells in vivo to improve the efficacy of tumor immunotherapies [
132]. Although the mechanisms underlying dysfunction in these cell subtypes may not be as well understood as for conventional T cells, several strategies emerge to enhance both the in vitro expansion of these cells and their therapeutic potential.
2.3. NK Cells
Natural killer (NK) cells can produce a vast array of cytokines/chemokines and are key players in immune responses against tumors and infected cells. NK cells can also directly regulate T-cell responses as well as modulate antigen-presenting cell activation. In solid tumors, NK cell secretion of CC-chemokine ligand 5 (CCL5), XC-chemokine ligand 1 (XCL1) and XCL2, and IFNγ can promote the recruitment of dendritic cells and further mediate their activation, which have been shown to improve patient outcome [
133,
134]. However, as with T cells, NK cells can become exhausted, and blockading TIGIT has demonstrated potential in preventing NK-cell dysfunction. Hence, a direct checkpoint blockade in NK cells may result in a more potent tumor-specific T-cell response in an NK cell-dependent manner [
135,
136].
Activated NK cells also upregulate many receptors, which can be shaped according to culture conditions and media supplementation with cytokines such as IL-2, IL-12, IL-15, IL-18, or IL-21 and Type I IFNs [
137,
138,
139,
140,
141,
142,
143,
144]. Hence, ex vivo modulation of NK-cell receptor expression has also been extensively investigated to overcome dysfunction. In general, PBMCs are first depleted for CD3
+ cells and enriched for CD56
+ cells, then cultured in medium containing IL-2 for up to two weeks [
140,
145]. This ex vivo stimulation induces NK-cell cytokine secretion, STAT3/AKT signaling, and upregulation of the NKG2D receptor [
146]. IL-2 can also enhance NK-cell response to IL-12 by increasing the expression of its receptor [
147]. IL-15 supplementation was later used to inhibit activation-induced cell death, and activate the mTOR pathway and stress-activated genes, which confer better anti-tumor capacity [
148,
149,
150]. However, continuous IL-15 signaling has been linked to functional NK-cell exhaustion by decreased fatty acid oxidation [
151]. Moreover, it was shown that IL-12-mediated IFNγ production of NK cells requires priming with IL-18, a cytokine also known to enhance IL-15-induced NK-cell proliferation [
152,
153]. Finally, IL-21 has been used to further increase NK-cell proliferation and effector functions, even though it can trigger apoptosis in vitro [
154,
155,
156,
157]. Other studies demonstrated that the duration of NK cell exposure to IL-21 is in fact critical [
158,
159]. Since NK cells can also express the inhibitory receptor PD-1, it has been found that a blockade of the PD-1/PD-L1 axis can improve NK cell-mediated immunity to tumors, and this response is indispensable for the full therapeutic effect of immunotherapy [
160].
Another issue to consider is the NK-cell expansion and functional status from heavily pretreated and thus immunocompromised patients, which are much poorer than for allogeneic NK cells [
161]. Other than cytokine supplementation, investigation on feeder cells required for in vitro culture is underway. In a phase I clinical trial (
clinicaltrials.gov #NCT02481934), autologous NK cells were activated by an engineered K562 cell-expressed membrane-bound form of IL-15 and 4-1BB ligand [
162]. NK cells can also be derived from umbilical cord blood as an allogenic cell source (
clinicaltrials.gov #NCT01729091) (C).
2.4. Tumor-Infiltrating Lymphocytes
Tumor-infiltrating lymphocytes (TIL) are composed of antigen-experienced and “passenger” T cells found at the tumor site. The tumor reactive T cells are subjected to repeated antigen encounters. When harvested for immunotherapy purposes, these cells already exhibit signs of dysfunction that may in fact identify the cancer-reactive T cells [
163]. Still, they can be expanded prior to re-infusion into the patient. Although standard protocols use anti-CD3 stimulation with IL-2, generating T cells with a more advanced differentiation state, some groups focused on a cytokine combination cocktail during the expansion phase to increase cell functionality or on the selection of less differentiated TILs among the tumor [
23,
164,
165,
166].
TILs found in solid tumors indeed represent a heterogeneous population. It was recently discovered that TILs can be divided into two functionally distinct subsets [
167]. The first is the most abundant and is constituted of a clonally related terminally differentiated population that expresses high levels of inhibitory receptors. The other is a TCF1
+ stem-like CD8
+ T-cell population, which is suggested to be a major factor in the success or failure to eradicate a tumor depending on their ability to be sufficiently stimulated by an antigen-presenting-cell niche and to continuously produce terminally differentiated CD8
+ T cells within the tumor [
167].
It has also been found that the presence of the integrin αEβ7 (CD103), characteristic of tissue-resident memory T cells (Trm), is positively associated with cytokine production, whereas expression of the transcription factor EOMES is negatively associated with TIL function, suggesting a competition between an antitumor CD103+ Trm-like and an exhaustion program [
168]. CD69
+CD103
+ Trm cells usually reside in non-lymphoid tissues and function as a first line of defense against secondary infections. In addition to their unique anatomic location, Trm have distinct transcriptional profiles showing the upregulation of inhibitory receptors such as PD-1. However, human Trm have an enhanced capacity for production of certain cytokines and regulatory molecules and a decreased turnover compared to circulating effector memory T cells, suggesting long-term maintenance in situ [
169]. Furthermore, it has been demonstrated that the transcription factor BHLHE40 is specifically required for both Trm and TIL development as well as their polyfunctionality by sustaining mitochondrial fitness and a functional epigenetic state. Local PD-1 signaling in the tumor microenvironment inhibits TIL BHLHE40 expression, and BHLHE40 is critical for TIL reinvigoration following anti-PD-L1 blockade [
170].
Another approach to improve TIL fitness is to target the member of the tumor necrosis factor receptor superfamily T-cell co-stimulatory receptor 4-1BB (CD137). As 4-1BB is frequently present on non-exhausted CD8
+ TILs, a 4-1BB agonist and a PD-1 blockade demonstrated a synergistic survival benefit in a CD8
+ T-cell dependent manner. As such, combined treatment decreased TIL exhaustion and improved TIL functionality in a glioblastoma model [
171]. Similarly, a PD-1 blockade and 4-1BB stimulation were demonstrated to be an effective strategy to improve pancreatic tumor-reactive TIL yield [
172].
Following tumor infiltration, T cells interact with other immune, cancer, and stromal cells within the tumor microenvironment (TME). Cells comprised in this environment may develop premature senescence caused by external factors such as TME metabolic changes or drug and radiation therapy [
173]. Senescent cells stay metabolically active but cease to proliferate [
174,
175]. Another important characteristic of senescent cells is that the expression of many genes changes during senescence and gives rise to what is called the senescence-associated secretory phenotype (SASP) [
176]. Senescent cells secrete numerous biologically active factors, including cytokines, chemokines, growth factors, and proteases [
177,
178]. Because these secreted factors act in autocrine and paracrine manners and have pleiotropic effects on surrounding cells, they may virtually affect any cell type within the tumor microenvironment, including infiltrating T cells. Indeed, tumor-induced senescence in T cells can be reproduced in vitro by briefly incubating cells in conditions of a low tumor-to-T-cell ratio. Furthermore, senescent T lymphocytes become able to suppress the proliferation of normal T cells and promote tumor immune evasion [
179]. In anti-CD3 + IL-2 stimulated T cells, the inhibition of p38MAPK signaling proved to be helpful in reversing the senescence phenotype of CD8
+ T cells by increasing their proliferation and functionality [
48,
49] (D).
2.5. T-Cell Receptor (TCR) Transgenic Cells
Genetic engineering offers several possibilities, such as conferring new antigenic specificities to T cells and circumventing certain limitations linked to T-cell dysfunction. These approaches provide many advantages, as they minimise culture duration and offer the possibility of directly modulating key signaling pathways within engineered cells to reduce dysfunction. Among these strategies is the introduction of an artificial TCR into antigen-specific conventional T cells. Although it can confer more potent antitumor activity [
180], TCR gene transfer is fraught with the risk of inappropriate pairing between exogenous and endogenous TCR chains. Several strategies have thus been used to avoid mispairing that can result in suboptimal activity and novel off-target antigen reactivity, potentially leading to harmful immune side effects [
181]. Moreover, the transduced TCR must successfully compete with the endogenous TCR chains to form a ternary complex with the CD3 signaling complex [
182]. Hence, the native TCRβ gene, or simultaneous TCRα/β genes, have been knocked out using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 technology prior to transduction with a cancer-specific receptor of choice, resulting in a stronger and more polyfunctional response in engineered T cells when tested against target cancer cell lines [
183,
184]. Remarkably, the combination of CRISPR/Cas9 and TCR transgenic therapy has recently been used to knock out both the endogenous TCR chains and the negative co-signaling receptor PD-1 to redirected T cells bearing a transgenic TCR targeting a TAA from NY-ESO-1 [
185] (A).
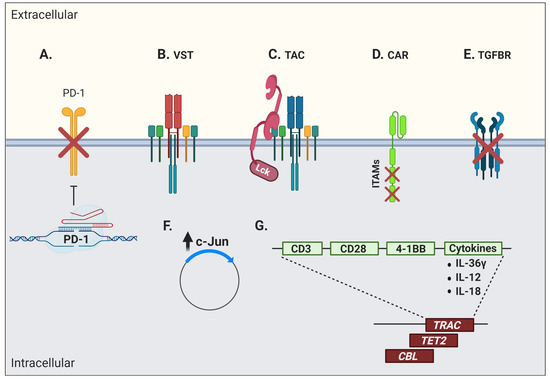
Figure 3. Interventions aiming at limiting T-cell dysfunction in engineered cells for adoptive immunotherapy. Engineered cells comprise transgenic T-cell receptor (TCR) and chimeric/synthetic antigen receptor T cells. (
A) Genetic ablation of negative co-signaling molecules (e.g., PD-1) may be used to circumvent the effects of T-cell dysfunction. (
B) Virus-specific T cells (VSTs) have been evaluated as a platform for artificial receptor expression in order to leverage their robust memory differentiation. (
C) Another strategy to increase modified T-cell fitness is the use of a T-cell antigen coupler (TAC) to avoid tonic signaling and leverage endogenous TCR signaling. (
D) Advanced engineering of CAR molecules involving the inactivation of two out of three ITAM motifs or (
E) the blockade of suppressive cytokine signaling, here shown by the overexpression of a dominant negative TGFβ receptor, can also promote better T-cell functions. (
F) An increase in c-Jun activity has been shown to further limit tonic signaling and T-cell dysfunction. (
G) Disruption of the endogenous TCRα/β chains by CRISPR/Cas9 or specific integration of the CAR vector into the targeted DNA locus (e.g., TRAC) have shown additional efficacy. This was also true when CAR constructs inadvertently disrupted the
TET2 and
CBL genes. Other strategies include the enhancement of T-cell function through the production of stimulatory cytokines (e.g., IL-36γ, IL-12, and IL-18) or co-stimulatory molecules (e.g., CD28 and 4-1BB). This figure was created with
BioRender.com.
The introduction of a transgenic TCR into virus-specific T cells (VSTs) to redirect their specificity towards cancer antigens has also been investigated [
186,
187,
188,
189,
190] (B). However, expression of a transgenic TCR often results in the downregulation of the endogenous TCR, which unfortunately leads to a reduced anti-viral reactivity [
186,
187,
188,
189,
190,
191]. This is in part explained by the competition for TCR signaling components by the endogenous and exogenous TCRs, and as possible consequences, one may expect lessened control of viral reactivations post-transplantation and poor capacity of TCR-transgenic VSTs to re-expand in vivo upon viral reactivation or vaccination [
190,
191]. The addition of CD8αβ to the transgenic TCR vector has been used to rescue endogenous MHC class I-restricted anti-viral TCR function [
192,
193]. These TCR-transgenic VSTs have a predominant central-memory phenotype and their anti-viral reactivity is preserved together with their anti-tumor function [
194]. The insertion of the CD8αβ co-receptor also improved antigen recognition by the TCR/MHC complex, recruitment of the tyrosine kinase Lck to the immune synapse, and proper activation of signaling components for T-cell activation, in addition to allowing CD4
+ T cells to recognize MHC class I-restricted antigens [
195,
196].
Another strategy to redirect T cells in a TCR-dependent, MHC-independent manner has been the use of a T-cell antigen coupler (TAC) composed of an antigen-binding domain, a TCR-recruitment domain, and a co-receptor domain (C) [
197]. This design recapitulates the architecture of a TCR complex and engages natural cellular pathways. In addition, these modified cells do not show signs of tonic signaling and display a less differentiated phenotype, which results in a potent T-cell product. These TAC-T cells also showed efficient tumor tissue infiltration at early time points post-ACT and great anti-tumor efficacy in a pre-clinical in vivo model of a solid tumor [
197]. Along the same lines, another group fused TCR subunits to an antibody-based binding domain to reprogram T-cell specificity in an HLA-independent manner while still taking advantage of the endogenous TCR signaling. These cells showed tumor cell killing in vivo, although they were less efficient in cytokine production, suggesting some degree of dysfunctionality or impaired activation [
198].
2.6. Chimeric Antigen Receptor (CAR) T Cells
Engineered T cells for the expression of an artificial receptor emerged in 1989 [
199]. This first-generation chimeric antigen receptor (CAR) T cell, composed of a CD3ζ chain containing three ITAMs for TCR-like signal transduction fused with single chain fragment of variable region (scFv) antibody, could support T-cell activation and cytotoxicity, but with very limited persistence and in vivo antitumor efficacy [
5,
200,
201]. Second-generation CARs therefore incorporated the two-signal model of T-cell activation by modifying CAR vectors to include a CD28 or 4-1BB co-stimulatory domain providing signals for T-cell effector function, proliferation, and more importantly, persistence [
202,
203]. Although complicated by massive activation and cytokine release syndrome, second-generation CD19-targeting CAR T cells rapidly entered routine clinical care. Third-generation CAR T cells further incorporate more than one co-stimulatory domain and other modifications or include the inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities [
204]. However, lack of CAR T-cell long-term persistence, poor expansion after ACT, and tumor immune escape remain cardinal limitations of this form of therapy [
205]. Hence, several issues pertaining to intrinsic biologic T-cell defaults impact the outcome of CAR-based treatments. Tonic CAR CD3ζ phosphorylation, triggered by antigen-independent clustering of scFv, has been shown to induce early CAR T-cell exhaustion. Moreover, integration of CD28 co-stimulation into the CAR vector seems to increase, whereas 4-1BB co-stimulation limits exhaustion induced by persistent CAR signaling [
206]. Indeed, stimulation of CD28/CD3ζ CARs activates faster with larger-magnitude changes in protein phosphorylation, which correlates with an effector T-cell phenotype. In contrast, 4-1BB/CD3ζ CAR T cells preferentially express T-cell memory-associated genes and exhibit sustained antitumor activity against established tumors in vivo [
207]. Another way to address the issue of tonic signaling is to calibrate the number of ITAMs on the CD3 moiety. Although the presence of one, two, or three functional ITAMs does not impede in vitro function, a single ITAM-containing CAR can outperform the other forms in vivo (D). Remarkably, this modified vector also favors persistence of highly functional CAR T cells, inducing long-lived memory cells with effective anti-tumor properties [
208].
Beyond concerns pertaining to T-cell engineering, the quality of the “input” material at the beginning of the manufacturing process impacts clinical outcomes. Autologous CAR T-cell efficacy greatly depends on the functional capacity of patients’ endogenous T cells. Indeed, studies have shown that T-cell fitness diminishes throughout the progression of diseases such as chronic lymphocytic leukemia (CLL), implying impaired proliferative capacity, a dysfunctional phenotype, and a decreased T-cell cytotoxicity, which impact the generation of CAR T cells [
209,
210,
211,
212,
213,
214]. Furthermore, molecular and functional T-cell defects are also acquired by a co-culture of previously healthy T cells with CLL cells [
213,
215,
216]. Among these defects, impairment of mitochondrial biogenesis and fitness, accompanied by reduced glucose transporter 1 (GLUT1) reserves, has been identified, which negatively correlates with the persistence of the transferred CAR T cells and clinical outcome [
217]. It was further shown that the frequency of memory T cells, defined by a CD8+CD45RO-CD27+ population, in the pre-manufacturing leukapheresis product was significantly associated with clinical response [
218,
219]. Similarly, cell features and the magnitude of ex vivo expansion when harvested after a response to induction therapy to manufacture B-cell maturation antigen (BCMA)-specific CAR T cells would be expected to be more clinically effective compared to a leukapheresis product from relapsed/refractory multiple myeloma [
220]. Finally, despite the generation of a lesser number of cells, the beneficial effects of reduced culture duration manifests in improved in vitro proliferation and effector function, which directly correlates with improved engraftment and anti-tumor function in vivo, even at a six-fold lower dose [
221].
The blockade of immunosuppressive pathways has also been investigated to confer superior functionality in CAR T cells. As such, it was demonstrated that exposure to TGFβ impairs proliferation as well as cytokine production of receptor tyrosine kinase-like orphan receptor 1 (ROR1)-specific CAR T cells co-cultured with ROR1-expressing triple-negative breast cancer cells. Thus, the blocking of TGFβ receptor signaling with a specific kinase inhibitor could promote better anti-tumor function in vitro [
222]. TGF-β is abundant in several cancer microenvironments (secreted by the tumor cells themselves, stromal cells, or infiltrating immunosuppressive cells such as myeloid derived suppressor cells—MDSC) and is a prime target for enhancing ACT. However, this is to be undertaken with caution, as TGFβ can also be beneficial for T-cell memory differentiation in certain contexts [
223]. After adoptive transfer, the overexpression of a dominant negative TGFβ receptor or hybrid receptors converting an inhibitory signal (such as PD-1) into a stimulatory signal are additional strategies to enhance ACT efficacy (E) [
224,
225]. Along the same lines, genetic ablation of negative co-signaling molecules such as PD-1 in CAR T cells, or CAR T cells secreting anti-PD-1 antibodies, is currently being investigated in clinical trials (
clinicaltrials.gov #NCT04213469, #NCT04489862) (A).
Studies on CAR metabolic pathway activation revealed that AP-1, a transcription factor composed of dimers of c-Jun and c-Fos, was highly solicited. Its activity is regulated by extracellular signals that can repress or activate its transcription. The formation of the AP-1 complex downstream of the TCR signaling induces IL-2 transcription, among other factors [
226,
227]. CAR T-cell exhaustion has also been associated with a profound defect in the production of IL-2, along with increased chromatin accessibility of AP-1 transcription factor motifs and overexpression of the basic leucine zipper (bZIP) and IRF transcription factors [
33,
228,
229]. Thus, overexpression of c-Jun rendered CAR T cells resistant to exhaustion, enhanced their expansion potential, increased their functional capacity, and diminished their terminal differentiation [
230] (F).
Fourth-generation CARs, also called “T cells redirected for antigen-unrestricted cytokine-initiated killing” or TRUCKs, are armored to improve cell fitness by inserting genes coding for other molecules such as cytokines, into the CAR vector [
231] (G). Interleukin-36γ (IL-36γ), for instance, showed significantly improved CAR T-cell expansion and persistence, and resulted in superior tumor eradication compared to conventional CAR T cells. The enhanced cellular function by IL-36γ was mediated in an autocrine manner. Furthermore, the activation of endogenous antigen-presenting and T cells by IL-36γ promoted a secondary anti-tumor response, which delayed the progression of an antigen-negative tumor challenge [
232]. IL-18-armored CAR T cells have also demonstrated enhanced proliferation and persistence in pre-clinical models, in addition to inducing a broadened anti-tumor response through endogenous immune effectors [
233,
234,
235]. Similarly, a CAR T cell secreting the pro-inflammatory cytokine IL-12 demonstrated an improved cytotoxicity and the ability to overcome an immune inhibitory microenvironment in several models, but a clinical trial using IL-12-secreting TILs revealed the high risk of toxicity of this approach if IL-12 secretion is not limited in time and space [
236,
237]. An attractive strategy to limit cytokine secretion into activated T cells is to use a NFAT-inducible system [
238].
As seen with conventional T cells, the use of endogenous TCR signaling has been investigated to improve the expansion and function of CAR T cells. As such, virus-specific T cells were modified with a CD19-specific CAR vector and infused into patients without prior cytoreductive chemotherapy. This approach is attractive for two reasons: It leverages the qualities of long-lived memory cells and can use viral reactivation as an adjuvant. In patients with viral reactivation, a striking proliferation of CAR T cells was observed with an associated depletion of CD19-expressing B cells, suggesting that dual TCR and CAR stimulation can potentiate engineered cell expansion [
239].
Since CAR constructs are usually generated with the use of viral vectors, integration may result in adverse effects, such as oncogenic transformation or uncontrolled growth, transgene expression, or transcriptional silencing [
240]. In a case report of CD19-specific CAR T-cell infusion for CLL treatment, an impressive clinical response was associated with the persistence of one major T-cell clone. It was further shown that the random integration of the CAR vector had disrupted the methylcytosine dioxygenase
TET2 gene, which led to an epigenetic profile consistent with altered T-cell differentiation and a central memory phenotype [
241]. In a case of anti-CD22 CAR T-cell therapy to treat B-cell acute lymphoblastic leukemia (ALL), a dominant T-cell clone containing a copy of the vector integrated into the second intron of the E3 ubiquitin-protein ligase
CBL gene was discovered. A loss of CBL has been associated with a reduction in the threshold for T-cell activation and dependence on co-stimulation [
242]. As such, integration of the CAR vector in this region resulted in a dominant-negative effect with the normal CBL (and/or its homolog CBL-B) function, thus contributing to the hyperexpansion in response to a small amount of antigen [
243]. Hence, attempts to direct the CAR vector into specific DNA regions are obviously of interest. Indeed, the targeted integration of a CD19-specific CAR vector into the T-cell receptor α constant (
TRAC) locus resulted in a more physiologic CAR expression, therefore limiting tonic CAR signaling and delaying effector T-cell differentiation and exhaustion [
244] (G).
Given that iPSCs can be easily amenable to genetic transformations in vitro, T-iPSCs can be genetically modified to augment their applicability, potency, and persistence and offer a great advantage compared to primary cells [
245]. Thus, antigen specificity could be assigned to T-iPSCs by means of a chimeric receptor [
246]. However, optimization is still needed, as these first iPSC-derived CAR-expressing T cells were still prone to more a terminally differentiated phenotype [
246].