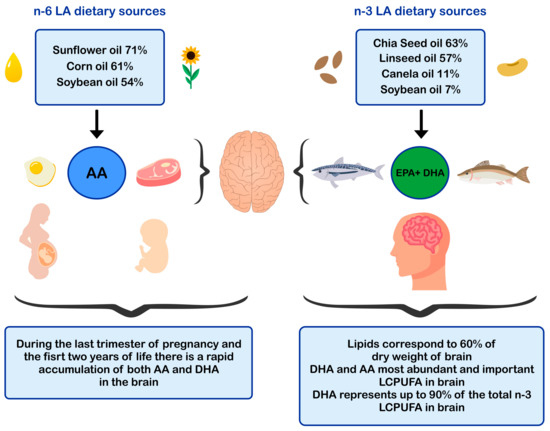
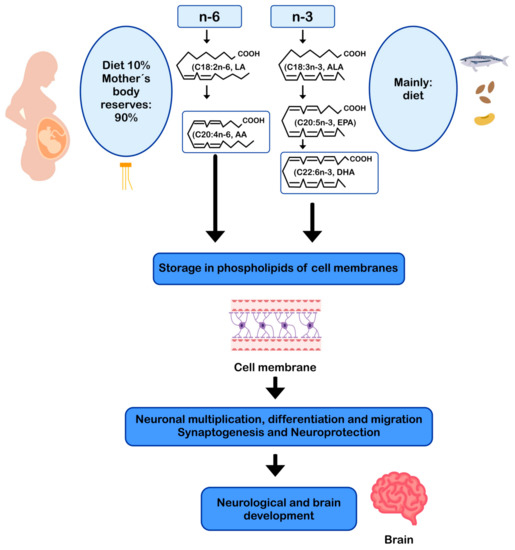
4. DHA and AA in Neuroprotection
The most frequent neurodegenerative age-related diseases are Alzheimer’s Disease (AD) and Parkinson’s Disease (PD) [
119]. Although the etiopathogenesis and clinical characteristics of AD and PD are different, these diseases share common mechanisms of damage, such as mitochondrial dysfunction [
120], neuroinflammation [
121], and oxidative stress [
122]. AD is characterized by dementia, memory loss, and cognitive decline, disorders that are worsened with aging [
123]. In 2019, the World Alzheimer Report indicated that more than 50 million people live with dementia worldwide, a number that will increase up to 152 million by 2050 [
124]. In this regard, every three seconds, a person develops dementia, and the current annual cost of dementia is estimated at US $ 1 trillion, which is estimated to double by 2030 [
124].
DHA via enzyme 15-lipoxygenase (15-LOX) can be converted in oxylipins, including resolvins and neuroprotectins, which are potent lipid mediators [
125,
126]. Furthermore, DHA may undergo lipid peroxidation producing oxylipin metabolites, such as 4-hytoxyhexenal (4-HHE) [
126,
127]. These metabolites may have a role in modulating oxidative cell homeostasis by regulating the activity of the transcription factors nuclear factor kappa-B (NF-κB) and nuclear factor erythroid 2-related factor 2 (Nrf2), thus participating in the inflammatory and antioxidant response, and neuroprotection [
126]. Osterman et al. (2019) [
128] assigned healthy adults with low fish consumption (
n = 121) to receive capsules with different doses of n-3 LCPUFA reflecting three patterns of fatty fish consumption: 1, 2, or 4 servings/week with 3.27 g of EPA + DHA (1:1.2) per serving or placebo. The authors reported that plasma oxylipins after 3 and 12 months increased linearly with the highest intake of EPA and DHA [
128].
Aging is a normal process of the life cycle, and its progression is usually accompanied by the decrease of a wide range of body functions, including cognitive function, marked by decreased synaptic density, decreased neuronal survival, and loss of volume of the gray and white matter [
105,
129,
130]. Alteration of lipid metabolism also occurs [
131] and is associated with the dysfunction of fluidity and activity of brain cell membranes microdomains (or rafts) [
132]. Exacerbation of alteration of lipid metabolism generates abnormal brain activity that can potentially lead to the development of neurodegenerative diseases [
132,
133]. Factors contributing to the early cognitive decline include diseases related to unhealthy lifestyles and metabolic syndrome [
104]. Among them, atherosclerosis and hypertension are relevant because an altered blood flow generates hypoperfusion and vascular dysfunction, causing an impairment of the blood-brain barrier and triggering neurodegenerative processes that lead to cognitive deterioration and, ultimately, to the development of AD [
131,
132].
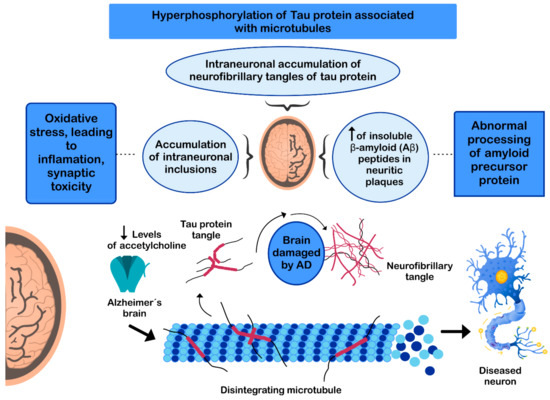
The pathogenesis of AD has not been fully resolved. However, it is known that the mechanisms that trigger the pathology are related to: (i) low levels of acetylcholine [
134]; (ii) aggregation of insoluble β-amyloid (Aβ) peptides, a product of abnormal processing of the amyloidal precursor protein, in neuritic plaques, leading to Aβ accumulation in the CNS [
135,
136,
137]; (iii) hyperphosphorylation of the tau protein associated with microtubules, causing intraneuronal accumulation of neurofibrillary tangles of tau protein and disruption of neuronal microtubules [
138,
139]; and (iv) oxidative stress, leading to inflammation, synaptic toxicity and accumulation of intraneuronal inclusions [
136,
140] (). Regarding its clinical characteristics, AD is a slowly progressive disease and three stages can be recognized in its evolution: the first is characterized by memory failures; in the second, language disorders, apraxias, and Gerstmann syndrome (agraphia and agnosia) are frequently added; and in the third stage, the patient is physically disabled and prostrated [
141,
142].
Figure 3. Mechanisms involved in the pathogenesis of Alzheimer’s disease (AD).
PD is the second most common neurodegenerative disease after AD [
143], affecting 2%-3% of the population ≥65 years old [
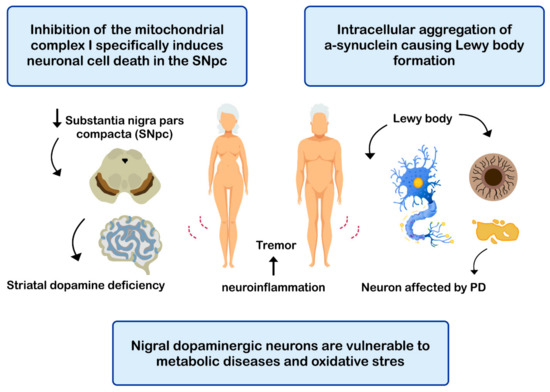
144]. The etiopathogenesis of the disease includes: (i) intracellular aggregation of α-synuclein causing the formation of Lewy bodies [
145,
146]; (ii) neuronal loss of the substantia nigra pars compacta (SNPc) leading to a marked striatal dopamine deficiency [
147,
148]; (iii) mitochondrial dysfunction, due to defects in the activity and incorrect assembly of complex I of the mitochondrial electron transport chain [
149,
150] (inhibition of mitochondrial complex I induces neuronal cell death in the SNPc leading to dopaminergic decrease) [
147,
149]; (iv) oxidative stress [
147], the nigral dopaminergic neurons are particularly vulnerable to metabolic diseases and oxidative stress [
151,
152,
153,
154]; and (v) neuroinflammation [
147,
155,
156] (). The clinical evolution of PD is characterized by motor disturbances due to progressive nigrostriatal dopaminergic neurodegeneration that occurs in parallel with decreased levels of striatal dopamine, dopaminergic synapses, and the density of dendritic spines in mid-striated spinal neurons [
157]. Other non-motor symptoms of PD include cognitive impairment [
158] and gastrointestinal dysfunctions [
159,
160].
Figure 4. Mechanisms related with the pathogenesis of Parkinson’s disease (PD).