Bile acids (BAs) are facial amphiphiles synthesized in the body of all vertebrates.
- bile acids
- physiological functions
- bile acid derivatives
- pharmacological application
1. Introduction
Bile acids (BAs) constitute an important class of biological molecules produced in the metabolism of all vertebrates. In mammals, they exhibit the so called C24 structure: 24 carbon atoms form a steroid nucleus (three six-member rings indicated as A, B C and a five-member ring indicated as D) and a five-carbon side chain with a carboxyl group at the C-24 position.
The A and B rings are linked in cis configuration, inducing an overall bent shape. Such a structural feature delineates a concave and convex side of the steroidal backbone where OH groups in α orientation (up to three) and two methyl groups in β orientation, respectively, point out. Therefore, two opposite faces with hydrophilic and hydrophobic properties can be distinguished. (Figure 1a,b). Further variations of the molecular structure can be observed at C-3 carbon due to hydroxyl, sulfate or glucuronate substituents [1][2]. C-6 and C-24 glucoronide conjugates were also found in humans [2]. Other C-24 substituents are glycine or taurine [3][4][5][6]. Recently Dorrestein et al. reported new amino acid C-24 substituted cholic acid (CA) namely phenylalanocholic, tyrosocholic and leucocholic acid [7]. BA actions generally occur in conditions where they are deprotonated; for this reason, many authors refer to them as bile salts instead of acids. In this review the term BA will be used keeping in mind that we refer mostly to their salt form.
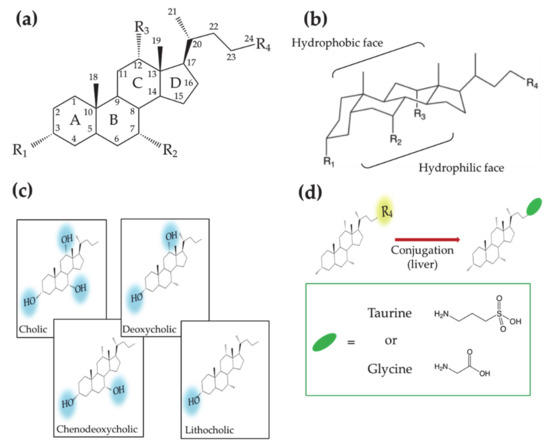
Figure 1. (a) Planar representation of the general molecular structure of bile acids (Bas). Letters, numbers and labels Ri indicate the rings of the steroid skeleton, the carbon atoms and the functional groups, respectively. (b) Chair representation of the general molecular structure of BAs. Brackets indicate the hydrophobic and hydrophilic faces. (c) Molecular structures of BAs showing different hydroxyl groups on the steroid backbone. (d) Molecular structures of the aminoacid conjugated BAs.
According to the order in which they are produced in the human body, BAs are differently named. CA and chenodeoxycholic (CDCA) acids—3 and 2 OH groups, respectively—are first synthesized by the hepatocyte and thus named primary BAs. Subsequently CA and CDCA are conjugated to glycine or taurine, giving rise to glycocholic (GCA), taurocholic (TCA) acids and glycochenodeoxycholic (GCDCA), taurochenodeoxycholic (TCDCA) acids, respectively. Further metabolization leads to the secondary BAs, deoxycholic (DCA) and lithocholic (LCA) acids, that present two and one OH groups, respectively (Figure 1c,d). The increase in the hydrophobic character, moving from primary to secondary BAs, affects the BA chemical–physical and physiological properties, making them differently active in the diverse parts of the enterohepatic circuit.
Generally speaking, five major physiological roles can be distinguished in BA activity: (1) regulation of cholesterol homeostasis; (2) deterrence for the formation of gallstones and kidney stones; (3) emulsification of dietary lipids and absorption of fat-soluble vitamins; (4) antimicrobial activity; (5) regulation function.
These functions will be described in the next paragraphs, following the BA physiological pathway—from biosynthesis to elimination/recirculation.
BAs are produced in the liver and stored in the gallbladder. Subsequently they are secreted through the biliary tract in the intestine, absorbed in the intestinal epithelium to pass into the portal circulation and return to the liver. The overall process is named enterohepatic circulation [8]. A small part of BAs escapes this cycle being secreted through the feces. The daily loss of BAs is compensated by new liver production.
2. Synthesis in the Liver and Storage in the Gallbladder: BAs in Cholesterol Metabolism
BAs are produced in their primary forms (CA and CDCA) in the liver, adopting cholesterol as starting substrate and the activity of 16 enzymes. Such enzymes catalyze 17 different reactions. In humans, the synthesis takes place in multiple intracellular compartments such as the cytosol, endoplasmic reticulum, mitochondria and peroxisomes.
A living organism can exploit two different synthetic routes for the BA synthesis [9].
The route, that is quantitively more important for adult humans, is known as “classic or neutral path” and provides for more than 90% of the BA needs. Such a synthetic pathway starts with the hydroxylation of the sterol ring on C7 mediated by the cholesterol 7α-hydroxylase (CYP7A1). Subsequently the intermediate is immediately modified on the lateral chain by the sterol 27-hydroxylase (CYP27) for the CDCA synthesis or, in the case of the CA synthesis, is further hydroxylated on C-12 by the 12α-hydroxylase (CY8B1) before the CYP27 catalyzed step. In this pathway, the CYP7A1 activity is the kinetic key that determines the overall rate of the metabolic pathway. On the other hand, the CY8B1 activity, modulating the hydrophilicity composition of the steroid nucleus, controls the CA and CDCA production ratio.
The second biosynthetic route, named “acidic path”, leads to the production of CDCA in humans and it is dominant in human neonates. In this pathway the chemical modifications of the cholesterol substrate involve first the lateral chain where a hydroxyl group is introduced on C27 by the CYP27 activity. Subsequently the modification of the sterol part is performed by the oxysterol 7α-hydroxylase (CYP7B1). A minority fraction of C25- and C24-hydroxycholesterols generated by the corresponding hydroxylases can also enter the acidic pathway to form BAs. Although most of the hydroxycholesterols are produced in the liver, hydroxycholesterols generated in extra hepatic tissues, such as lungs and brain, may be transported to the liver and be also involved in this BAs biosynthetic route. After their synthesis, CA and CDCA are conjugated to taurine and glycine by the activity of two enzymes, bile acid-CoA synthase and bile acid-CoAamino acid N-acyltransferase. The conjugation makes the produced BA more hydrophilic and more acidic on the side chain: the pKa decreases from ~5.0 to ~3.9 and <2 for glycine and taurine conjugates, respectively. The amount of synthesized BA is regulated by control mechanisms that operate at transcriptional level, where the transcription factors are nuclear receptors. An exceeding amount of BA triggers a negative feedback mechanism, starting with the BAs binding to the hepatic farnesoid X receptor (FXR) and ending with the inhibition of the genes expressing the CYP7A1 and CYP8B1 enzymes involved in the biosynthesis of BA [10][11]. The activation of FXR affects not only the synthesis itself but it also regulates the level of the BA in the intestine and biliary tree. Indeed, in order to assure concurrently an efficient lipid absorption and a sustainable hepatic level of BA, the FXR increases the expression of the transporters which mediate the efflux of the BA into the biliary canaliculi. At the same time, it suppresses the expression of the importer NTCP, thus reducing the BA reabsorption from the blood into the hepatocytes [12]. Besides the effect in the liver, reabsorbed BAs can bind also the intestinal FXR in the distal ileum, activating it. The activated FXR stimulates in turn the expression of the fibroblast growth factor 19 (FGF19) and its release into the portal blood. Once it reaches the hepatocyte plasma membrane, FGF19 binds the FGF receptor 4 (FGFR4)/β-klotho complex, triggering a signaling cascade that results in the suppression of the CYP7A1 mRNA and thus the suppression of the CYP7A1 expression [13][14].
BA production utilizes a consistent amount of the cholesterol pool (about 500 mg per day), thus turning out to be one of the main mechanisms for cholesterol regulation in the human body. [15] Consistently, hypercholesterolemia is often treated by Bas sequestrants [16][17][18][19]. It has been observed that certain animals when fed with hypercholesterolic diet manage to keep normal the plasma cholesterol level, thanks to a compensating mechanism that increases the production of BAs [20]. BAs sequestrants [16] are generally formed by cationic polyelectrolytes, which are able to bind the BAs through both electrostatic and hydrophobic attractive interactions and as such to remove them from the enterohepatic circulation. Sequestration stimulation increased conversion of cholesterol into BAs in the liver, thereby leading to the lowering of the LDL (low-density lipoproteins, “bad”) cholesterol in the blood, and simultaneously to an increase in the HDL (high-density lipoproteins, “good”) cholesterol and triglycerides [21][22]. BA sequestrants are also used in the medical treatment of BA related diseases, such as BA diarrhea caused by BAs in the large intestine or colon due to the malabsorption of BA in the small intestine (secondary BA diarrhea) or BA overproduction resulting from defective feedback inhibition of the biosynthesis of BAs in the liver (primary BA diarrhea) [23][24][25][26]. In the treatment, sequestrants bind BAs, hinder them from contact with the colonic mucosa, thereby decreasing the BA level in the colon [27][28][29]. BA sequestrants can also reduce glucose levels in patients with type 2 diabetes mellitus, although the mechanism of action still remains unclear [30]. Moreover, BAs malabsorption reduces the amount of BA in the small intestine inducing fat maldigestion and consequent steatorrhea as occurring in patients with short bowel syndrome [31]. Consequently, the maldigested fatty acids in the small intestine complex the luminal Ca, lowering the fraction of Ca available for the dietary oxalate precipitation. In turn, the oxalate remains as free ions and is hyperabsorbed by the colon, leading to hyperoxaluria and increasing the possibility of kidney stone formation. Oral therapies based on natural and synthetic conjugated bile salts were observed to decrease fecal fat and urinary oxalate execration in patients with short bowel syndrome [32][33].
A further important role of BAs towards cholesterol concerns the solubilization activity in the bile. Cholesterol is majorly eliminated through the secretion in bile. Bile is a solution produced in the liver that is composed by 95% of water. Free cholesterol is insoluble in aqueous solutions. However, in bile due to the presence of BAs (about 0.7%) and lipids like lecithin, cholesterol is easily solubilized through the formation of mixed micelles. Such a process avoids the cholesterol supersaturation and in turn the formation of gallstones. The interaction between cholesterol and BAs has been analyzed both from a chemical and medical point of view. In the first case, several studies analyzed the structure, stability and parameter formation of BAs–cholesterol-based micelles and crystals [34][35][36]. In the latter case, CDCA was proved in the 1970s as successful molecule for dissolution of cholesterol gallstones [37][38][39][40]. However, its use was lately abandoned because of the occurrence of side effects. The more hydrophilic ursodeoxycholic acid (UDCA) and its taurine conjugate started to be investigated as alternative treatment for cholelithiasis and their use in oral therapy is still ongoing, albeit restricted to a specific target group of patients (e.g., patients having gallstones due to temporary and non-genetic causes) [41]. The combined efficiency of UDCA and polyunsaturated fatty acids in dissolving cholesterol gallstones in mice was recently reported by Lee et al. [42].
After the synthesis, conjugated BAs are transferred into bile, passing through the hepatocyte’s membrane to the canaliculi via the bile salt export pump. The bile in the canaliculi converge in a series of ducts that eventually terminate into the common hepatic duct. Via the hepatic duct, the bile reaches the gallbladder, where it is stored or is delivered directly to the intestinal lumen [43].
3. From the Gallbladder to the Intestine: Lipid Solubilization and Absorption
After a meal, the cholecystokinin hormone is released and its presence is the signal for the gallbladder to release bile. At this point the aminoacidic conjugation is functional for the BA activities since it allows for the transits to the small intestine via the biliary tree. Indeed, being completely ionized at the pH of biliary tract and small intestine, conjugated BAs cannot diffuse through the cell membranes, thus assuring consistent intraluminal concentration in the intestine for lipid digestion. Moreover, the conjugated BAs are less prone than unconjugated BAs to precipitate in presence of high concentration of Ca2+ ions. In the distal small intestine, bacteria break the conjugation with the aminoacidic of a fraction of conjugated BAs. Deconjugation is completed in the colon where further modifications as dihydroxylation and epimerization occur to give rise to secondary BAs, i.e., DCA, LCA and UDCA. Recently, anaerobic in vitro reconstitution experiments showed that six enzymes are sufficient for the conversion of cholic acid into DCA [44].
At any time, a portion of 85–90% of BAs is present in the small intestine. Here BAs have several roles in the digestion of lipids, ranging from emulsification to transport (Figure 2a) [45][46]. Lipid digestion starts in the stomach where the food is initially broken down by the mechanical action of peristalsis and by the chemical activity of gastric juices. Subsequently the digestion continues in the duodenum where the partially digested food mixes with digestive enzymes from the pancreas—pancreatic lipase—and BAs. At this stage, the ingested lipids are in the form of oil-in-water emulsion stabilized by different surface-active substances such as proteins and phospholipids. In order to hydrolyze lipids into simpler and absorbable molecules, lipase and its co-factor co-lipase have to anchor on the droplet surface. The first crucial role of BAs is to increase the bioavailability of the lipid substrates to the enzyme by displacing the different stabilizers at the water–oil interface. Subsequently BAs indirectly help the positioning of lipase on the lipidic substrate by favoring the interfacial adsorption of co-lipase. After the lipase activity, lipids are decomposed in free fatty acids and monoglycerides. These products remain at the oil–water droplet interface until BAs englobe them in mixed micelles, thereby providing their removal and transport through the intestinal mucus.
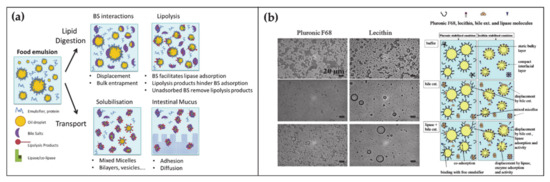
Figure 2. (a) Schematic representation of the BA functions and self-assembly during the lipid digestion and transport in the intestine (adapted from Macierzanka, A.; Torcello-Gómez, A.; Jungnickel, C.; Maldonado-Valderrama, J. Bile Salts in Digestion and Transport of Lipids. Adv. Colloid Interface Sci. 2019, 274, 102045. Ref. [46] with permission from (2019) Elsevier). (b) Transmission Electron Microscopy images of oil-in-water emulsions stabilized by two surfactants with interest in food and drug industry, namely Pluronic F68 and Lecithin (left top). Transmission Electron Microscopy images of the surfactant-emulsion transformation upon BA (left center) and BA + lipase addition (left bottom). Scheme representing the disposition of surfactant-BA-lipase at the oil/water interface (right). Reproduced from Torcello-Gómez, A.; Maldonado-Valderrama, J.; Martín-Rodríguez, A.; McClements, D.J. Physicochemical Properties and Digestibility of Emulsified Lipids in Simulated Intestinal Fluids: Influence of Interfacial Characteristics. Soft Matter 2011, 7, 6167–6177. Ref. [47] with permission from The Royal Society of Chemistry.
The fundamental understanding of these mechanisms is supported by a large collection of literature that by means of microscopic, spectroscopic and rheological techniques, (i) analyzed the displacement mechanism of BAs with respect to a large number of proteins and lipids (Figure 2b), (ii) demonstrated the improved activity of lipase in presence of BAs, (iii) elucidated the mechanism of lipid transport through the intestinal mucus [45][48][49][50]. Besides clarifying the physiological mechanism, such knowledges turn out to be essential for food and drug industry in order to engineer efficient strategies for drugs, dietary lipids and sugars uptake in the gastrointestinal tract [51][52][53][47].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22041780
References
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting Bile-Acid Signalling for Metabolic Diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693.
- Barbier, O.; Trottier, J.; Kaeding, J.; Caron, P.; Verreault, M. Lipid-Activated Transcription Factors Control Bile Acid Glucuronidation. Mol. Cell. Biochem. 2009, 326, 3–8.
- Kuhajda, K.; Kandrac, J.; Kevresan, S.; Mikov, M.; Fawcett, J.P. Structure and Origin of Bile Acids: An Overview. Eur. J. Drug Metab. Pharm. 2006, 31, 135–143.
- Madenci, D.; Egelhaaf, S.U. Self-Assembly in Aqueous Bile Salt Solution. Curr. Opin. Colloid Interface Sci. 2010, 15, 109–115.
- Galantini, L.; di Gregorio, M.C.; Gubitosi, M.; Travaglini, L.; Vázquez, J.; Jover, A.; Meijide, F.; Soto, V.H.; Pavel, N.V. Bile Salts and Derivatives: Rigid Unconventional Amphiphiles as Dispersants, Carriers and Superstructure Building Blocks. Curr. Opin. Colloid Interface Sci. 2015, 20, 170–182.
- di Gregorio, M.C.; Travaglini, L.; Del Giudice, A.; Cautela, J.; Pavel, N.V.; Galantini, L. Bile Salts: Natural Surfactants and Precursors of a Broad Family of Complex Amphiphiles. Langmuir 2019, 35, 6803–6821.
- Quinn, R.A.; Melnik, A.V.; Vrbanac, A.; Fu, T.; Patras, K.A.; Christy, M.P.; Bodai, Z.; Belda-Ferre, P.; Tripathi, A.; Chung, L.K.; et al. Global Chemical Effects of the Microbiome Include New Bile-Acid Conjugations. Nature 2020, 579, 123–129.
- Roberts, M.S.; Magnusson, B.M.; Burczynski, F.J.; Weiss, M. Enterohepatic Circulation. Clin. Pharm. 2002, 41, 751–790.
- Russell, D.W. Fifty Years of Advances in Bile Acid Synthesis and Metabolism. J. Lipid Res. 2009, 50, 120–125.
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Mol. Cell 2000, 6, 517–526.
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptos. Mol. Cell 2000, 6, 507–515.
- Denson, L.A.; Sturm, E.; Echevarria, W.; Zimmerman, T.L.; Makishima, M.; Mangelsdorf, D.J.; Karpen, S.J. The Orphan Nuclear Receptor, Shp, Mediates Bile Acid-Induced Inhibition of the Rat Bile Acid Transporter, Ntcp. Gastroenterology 2001, 121, 140–147.
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential Regulation of Bile Acid Homeostasis by the Farnesoid X Receptor in Liver and Intestine. J. Lipid Res. 2007, 48, 2664–2672.
- Kong, B.; Wang, L.; Chiang, J.Y.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of Tissue-Specific Farnesoid X Recptor in Suppressing the Expression of Genes in Bile-Acid Synthesis in Mice. Hepatology 2012, 56, 1034–1043.
- Li, T.; Francl, J.M.; Boehme, S.; Chiang, J.Y.L. Regulation of Cholesterol and Bile Acid Homeostasis by the Cholesterol 7α-Hydroxylase/Steroid Response Element-Binding Protein 2/MicroRNA-33a Axis in Mice. Hepatology 2013, 58, 1111–1121.
- Meissner, M.; Wolters, H.; de Boer, R.A.; Havinga, R.; Boverhof, R.; Bloks, V.W.; Kuipers, F.; Groen, A.K. Bile Acid Sequestration Normalizes Plasma Cholesterol and Reduces Atherosclerosis in Hypercholesterolemic Mice. No Additional Effect of Physical Activity. Atherosclerosis 2013, 228, 117–123.
- Mazidi, M.; Rezaie, P.; Karimi, E.; Kengne, A.P. The effects of bile acid sequestrants on lipid profile and blood glucose concentrations: A systematic review and meta-analysis of randomized controlled trials. Int. J. Cardiol. 2017, 227, 850–857.
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—Bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498.
- Hou, R.; Goldberg, A.C. Lowering Low-Density Lipoprotein Cholesterol:Statins, Ezetimibe, Bile Acid Sequestrants, and Combinations: Comparative Efficacy and Safety. Endocrinol. Metab. Clin. N. Am. 2009, 38, 79–97.
- Sehayek, E.; Ono, J.G.; Shefer, S.; Nguyen, L.B.; Wang, N.; Batta, A.K.; Salen, G.; Smith, J.D.; Tall, A.R.; Breslow, J.L. Biliary Cholesterol Excretion: A Novel Mechanism That Regulates Dietary Cholesterol Absorption. Proc. Natl. Acad. Sci. USA 1998, 95, 10194–10199.
- Mandeville, W.; Goldberg, D. The Sequestration of Bile Acids, a Non-Absorbed Method for Cholesterol Reduction: A Review. Curr. Pharm. Des. 1997, 3, 15–28.
- Camilleri, M.; Gores, G.J. Therapeutic Targeting of Bile Acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 55905, 209–215.
- Lundasen, T.; Galman, C.; Angelini, B.; Rudling, M. Circulating Intestinal Fibroblast Growth Factor 19 Has a Pronounced Diurnal Variation and Modulates Hepatic Bile Acid Synthesis in Man. J. Intern. Med. 2006, 260, 530–536.
- Walters, J.R.F.; Tasleem, A.L.I.M.; Omer, O.S.; Brydon, W.G.; Dew, T.; Roux, C.W.L.E. A New Mechanism for Bile Acid Diarrhea: Defective Feedback Inhibition of Bile Acid Biosynthesis. Clin. Gastroenterol. Hepatol. 2009, 7, 1189–1194.
- Johnston, I.; Nolan, J.; Pattni, S.S.; Walters, J.R.F. New Insights into Bile Acid Malabsorption. Curr. Gastroenterol. Rep. 2011, 13, 418–425.
- Walters, J.R.F. Bile Acid Diarrhoea and FGF19: New Views. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 426–434.
- Hofmann, A.F.; Mysels, K.J. Bile Salts as Biological Surfactants. Colloids Surf. 1988, 30, 145–173.
- Huval, C.C.; Holmes-Farley, S.R.; Mandeville, W.H.; Sacchiero, R.; Dhal, P.K. Syntheses of Hydrophobically Modified Cationic Hydrogels by Copolymerization of Alkyl Substituted Diallylamine Monomers and Their Use as Bile Acid Sequestrants. Eur. Polym. J. 2004, 40, 693–701.
- Wilcox, C.; Turner, J.; Green, J. Systematic Review: The Management of Chronic Diarrhoea Due to Bile Acid Malabsorption. Aliment. Pharm. Ther. 2014, 39, 923–939.
- Hansen, M.; Sonne, D.P.; Knop, F.K. Bile Acid Sequestrants: Glucose-Lowering Mechanisms and Efficacy in Type 2 Diabetes. Curr. Diab. Rep. 2014, 14, 482.
- Seetharam, P.; Rodrigues, G. Short Bowel Syndrome: A Review of Management Options. Saudi J. Gastroenterol. 2011, 17, 229–235.
- Emmett, M.; Guirl, M.J.; Santa Ana, C.A.; Porter, J.L.; Neimark, S.; Hofmann, A.F.; Fordtran, J.S. Conjugated Bile Acid Replacement Therapy Reduces Urinary Oxalate Excretion in Short Bowel Syndrome. Am. J. Kidney Dis. 2003, 41, 230–237.
- Gruy-Kapral, C.; Little, K.H.; Fordtran, J.S.; Meziere, T.L.; Hagey, L.R.; Hofmann, A.F. Conjugated Bile ACid Replacement Theraphy for Short-Bowel Syndrome. Gastroenterology 1999, 116, 15–21.
- Van de Heijning, B.J.M.; Stolk, M.F.J.; Van Erpecum, K.J.; Renooij, W.; Groen, A.K.; VanBerge-Henegouwen, G.P. Bile Salt-Induced Cholesterol Crystal Formation from Model Bile Vesicles: A Time Course Study. J. Lipid Res. 1994, 35, 1002–1011.
- Tamesue, N.; Juniper, K. Concentrations of Bile Salts at the Critical Micellar Concentration of Human Gall Bladder Bile. Gastroenterology 1967, 52, 473–479.
- Higuchi, W.I.; Tzeng, C.S.; Chang, S.J.; Chiang, H.J.; Liu, C.L. Estimation of Cholesterol Solubilization by a Mixed Micelle Binding Model in Aqueous Tauroursodeoxycholate:Lecithin:Cholesterol Solutions. J. Pharm. Sci. 2008, 97, 340–349.
- Reynier, M.O.; Montet, J.C.; Gerolami, A.; Marteau, C.; Crotte, C.; Montet, A.M.; Mathieu, S. Comparative Effects of Cholic, Chenodeoxycholic, and Ursodeoxycholic Acids on Micellar Solubilization and Intestinal Absorption of Cholesterol. J. Lipid Res. 1981, 22, 467–473.
- Rudling, M.; Laskar, A.; Straniero, S. Gallbladder Bile Supersaturated with Cholesterol in Gallstone Patients Preferentially Develops from Shortage of Bile Acids. J. Lipid Res. 2019, 60, 498–505.
- Carey, M.C.; Small, D.M. The Physical Chemistry of Cholesterol Solubility in Bile. Relationship to Gallstone Formation and Dissolution in Man. J. Clin. Investig. 1978, 61, 998–1026.
- Coreta-Gomes, F.M.; Vaz, W.L.C.; Wasielewski, E.; Geraldes, C.F.G.; Moreno, M.J. Quantification of Cholesterol Solubilized in Dietary Micelles: Dependence on Human Bile Salt Variability and the Presence of Dietary Food Ingredients. Langmuir 2016, 32, 4564–4574.
- Portincasa, P. Therapy of Gallstone Disease: What It Was, What It Is, What It Will Be. World J. Gastrointest. Pharm. Ther. 2012, 3, 7–20.
- Jang, S.I.; Fang, S.; Kim, K.P.; Ko, Y.; Kim, H.; Oh, J.; Hong, G.Y.; Lee, S.Y.; Kim, J.M.; Noh, I.; et al. Combination Treatment with N-3 Polyunsaturated Fatty Acids and Ursodeoxycholic Acid Dissolves Cholesterol Gallstones in Mice. Sci. Rep. 2019, 9, 1–13.
- Boyer, J.L. Bile Formation and Secretion. Compr. Physiol. 2013, 3, 1035–1078.
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A Metabolic Pathway for Bile Acid Dehydroxylation by the Gut Microbiome. Nature 2020, 582, 566–570.
- Maldonado-Valderrama, J.; Wilde, P.; MacIerzanka, A.; MacKie, A. The Role of Bile Salts in Digestion. Adv. Colloid Interface Sci. 2011, 165, 36–46.
- Macierzanka, A.; Torcello-Gómez, A.; Jungnickel, C.; Maldonado-Valderrama, J. Bile Salts in Digestion and Transport of Lipids. Adv. Colloid Interface Sci. 2019, 274, 102045.
- Torcello-Gómez, A.; Maldonado-Valderrama, J.; Martín-Rodríguez, A.; McClements, D.J. Physicochemical Properties and Digestibility of Emulsified Lipids in Simulated Intestinal Fluids: Influence of Interfacial Characteristics. Soft Matter 2011, 7, 6167–6177.
- Naso, J.N.; Bellesi, F.A.; Pizones Ruiz-Henestrosa, V.M.; Pilosof, A.M.R. Studies on the Interactions between Bile Salts and Food Emulsifiers under in Vitro Duodenal Digestion Conditions to Evaluate Their Bile Salt Binding Potential. Colloids Surf. B. 2019, 174, 493–500.
- Cremers, C.M.; Knoefler, D.; Vitvitsky, V.; Banerjee, R.; Jakob, U. Bile Salts Act as Effective Protein-Unfolding Agents and Instigators of Disulfide Stress in Vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E1610–E1619.
- Pabois, O.; Lorenz, C.D.; Harvey, R.D.; Grillo, I.; Grundy, M.M.L.; Wilde, P.J.; Gerelli, Y.; Dreiss, C.A. Molecular Insights into the Behaviour of Bile Salts at Interfaces: A Key to Their Role in Lipid Digestion. J. Colloid Interface Sci. 2019, 556, 266–277.
- Moghimipour, E.; Ameri, A.; Handali, S. Absorption-Enhancing Effects of Bile Salts. Molecules 2015, 20, 14451–14473.
- Naumann, S.; Schweiggert-Weisz, U.; Eglmeier, J.; Haller, D.; Eisner, P. In Vitro Interactions of Dietary Fibre Enriched Food Ingredients with Primary and Secondary Bile Acids. Nutrients 2019, 11, 1424.
- Shokry, D.S.; Waters, L.J.; Parkes, G.M.B.; Mitchell, J.C.; Snowden, M.J. Formation of a Bile Salt-Drug Hydrogel to Predict Human Intestinal Absorption. J. Pharm. Sci. 2019, 108, 279–287.