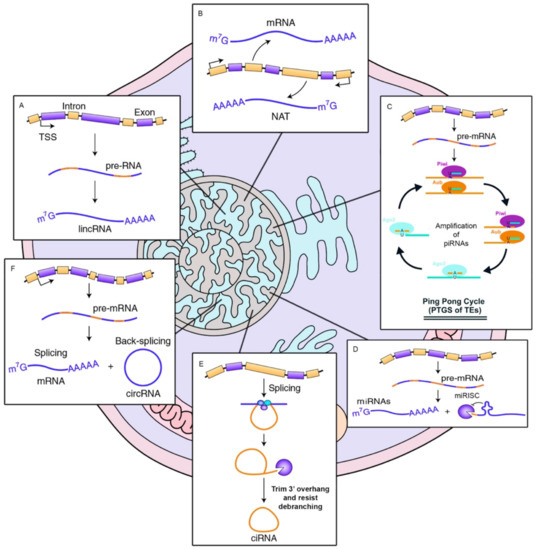
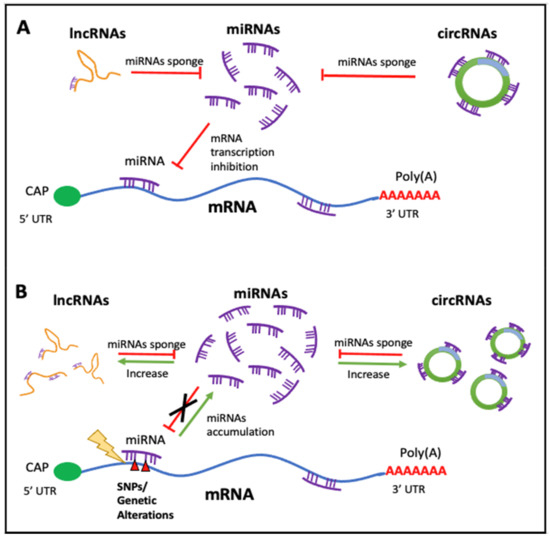
2.1. Non-Coding RNAs in Cancer
A large number of laboratories have published and experimentally validated that alteration in the expression profiles of certain non-coding RNAs are linked to development of cancer [
72]. In addition, single nucleotide polymorphisms (SNPs) are the genetic variations that are most frequently associated with the onset of cancer. Remarkably, 85% of SNPs are located in non-coding regions and linked to the development of this disease [
73].
In the case of miRNA and more specifically in cancer, there are the so-called OncomiRs, which appear overexpressed in tumor samples and linked to tumor development [
74]. It has been reported that OncomiRs affect the proliferation and signaling of cancer cells, prevent apoptosis, favors invasion, angiogenesis and metastasis [
75]. (Please see .) For example, the miR-17/92 cluster was the first OncomiR described. This is a polycistronic RNA that encodes for 6 miRNAs. Its overexpression is related to the inhibition of E2F1 [
76] and AIB1 genes [
77] triggering tumor initiation by stimulating cell progression and proliferation. On the contrary, anti-OncomiRs are a group of miRNAs that behaves as tumor suppressors, such as the miR-143/145 cluster. Both miRNAs control the expression of dozens of genes and its deregulation is believed to propitiate the appearance of the initial events in cancer. In comparison with cells from non-tumor tissues, these miRNAs are expressed in a smaller amount in colon, head and neck, breast, bladder, and lung cancers [
78].
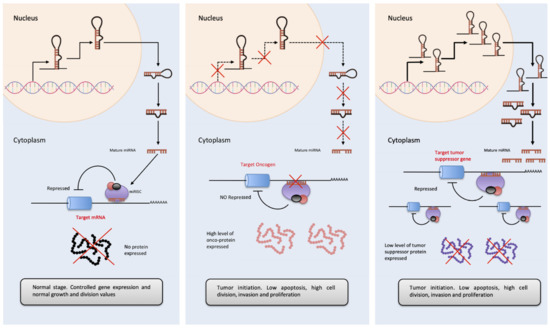
Figure 3. OncomiRs involvement in tumor initiation process. Left. The diagram represents basal expression of miRNAs. These are released to the cytoplasm in a highly controlled amount, to exert their function by binding to the 3′-UTR of their target mRNA, inhibiting the formation of protein. Center. In the case of cancer, the expression of multiple genes is altered, if this variation causes an inhibition of OncomiRs (miRNAs that regulate oncogene expressions), these are not able to inhibit the amount of oncogenic protein, favoring proliferation, cell cycle progression as well as a decrease in apoptosis and in the tumor initiation. Right. On the contrary, in cancer, the overexpression of miRNAs acting as tumor suppressor regulators has also been observed (essentially, they are miRNAs that inhibit the expression of tumor suppressor genes). A considerable increase in the cytoplasm of OncomiRs, causes the almost disappearance of tumor suppressor proteins, favoring the development of cancer, its progression and tumor invasion.
This indicates that a recurrent oncogenic process is implicated. Well known is the TP53 gene loss-of-function, as the activation of the p53 pathway increases the miR-143 and miR-145 levels through a transcriptional mechanism [
79]. Another mechanism that affects miR-143 and miR-145 levels depends on the mitogen-activated protein kinase (MAPK) cascade activity, with RREB1 being the effector for the repression of the cluster [
80]. In addition, in the case of colorectal cancer, the variant rs353292 in the flanking region of miR-143/145 increases the risk of developing this disease [
81], as rs353292 CT/TT individuals presented a lower expression of miR-143.
Furthermore, there is a group of miRNAs involved in an extensive range of diseases, those related to hypoxia (low oxygen levels) and called hypoxamirs. In the case of cancer, there are a high number of tumors with hypoxic tendency (being large masses of tissue with a high proliferation rate and little vascular development, the development of hypoxic regions is favored), such as breast cancer. Among this group of microRNAs, we must highlight miR-210, recognized as a mediator of cellular responses to stress related to hypoxia. Recent studies have postulated that hypoxamirs regulate hypoxic transcriptional cascades, angiogenesis, and endothelial growth [
82] and consequently, involved in metastasis. miRNAs that suffer an alteration in their expression levels, have been related to various functions in tumoral events, including the maintenance of proliferation, resistance to cell death, invasion, and metastasis and very recently with resistance to drugs [
83]. These anomalous expression patterns have been endorsed to DNA modifications (deletions, amplifications, or mutations) [
84], epigenetic alterations [
85], miss-regulated transcription factors [
86], and deregulation of RNA-binding proteins (RBP) that contribute to miRNA synthesis [
87].
In addition, single nucleotide polymorphisms (SNPs) are the genetic variations that are most frequently associated with the onset of cancer. Remarkably, 85% of SNPs are located in non-coding regions and linked to the development of this disease [
73]. For example, the SNP rs11671784 is localized within the miR-27a sequence affecting its processing efficiency. This variant has been implicated in gastric cancer reduction risk by impairing the maturation of pre-miR-27a to mature miR-27a [
88].
According to the atlas of human long non-coding RNAs in FANTOM5 [
89], there are about 28,000 known human lncRNA genes, making them the most abundant transcribed RNAs from DNA. They can work as enhancers, scaffolds or decoys by binding with RNAs or even with RNA binding proteins, which alters all cellular signaling regulation networks [
2]. For instance, the lncRNA MEG3 (maternally expressed gene 3) binds the p53 DNA binding domain, favoring the activation of P53 [
90]. Interestingly, in cervical cancer, MEG3 expression is inversely associated to tumor volume and metastasis, which indicates its strong role as a tumor suppressor through P53 and pointing out as a possible therapeutic target [
91]. Likewise, low levels of MEG3 have been linked with an increment in cell cycle progression and autophagy in bladder cancer [
92]. Moreover, another example of the role of the long non-coding RNAs in gene regulation is the lncRNA-PVT1 that usually acts as miRNA sponge towards the miR-200 family in normal breast tissue and loses its functionality in breast cancer cells, altering the genetic regulation in this tissue [
93].
A recent study proved that LINC00883 could act as miRNA “sponge” for miR-150 [
94] which promotes tumor cell proliferation by negatively regulating tumor suppressor gene SRCIN1 [
95] and Notch3 [
96]. In fact, Notch3 played an important role in oncogenesis and resistance to chemotherapy in lung cancer [
97]. Qi Sun et al. demonstrated that T allele of rs793544 was associated with the low expression level of LINC00883 in lung tumor tissues [
98]. Taken together, it seems that SNP rs793544 is related with the diminished expression level of LNC00883, increasing the risk of lung cancer by altering the expression of miR-150.
In the case of piRNAs, it was initially thought that their only function was related to the suppression of the activity of mobile elements (or transposons). In fact, the inhibition of piRNAs that normally suppresses the function of these elements can facilitate mutagenic retro-transpositions and DNA instability, thus favoring cancer initiation. Nevertheless, it is not an exclusive mechanism, because it has been proposed that the piRNA/PIWI complex can cause aberrant DNA methylation, which results in genomic silencing, stimulating the cell to enter in a “stem-like” stage [
99]. In detail, piRNAs can stimulate
cis methylation, in human lymphoma and breast cancer cell lines, single copy piRNAs favor methylation of gene-specific DNA through imperfect binding to genomic DNA [
100]. Moreover, piRNAs have been associated to cell cycle regulation, in fact, it has been discovered that alteration in the expression of piRNA/PIWI complex correlates with clinical variables in tumorigenic samples indicating a new role for piRNA in cancer. For instance, in breast cancer, four significantly regulated piRNAs have been discovered: piR-20365, piR-4987, piR-20582, and piR-20485 [
101]. In addition, genetic association studies have identified variants associated with cancer in piRNA loci. As an example, the SNP rs147061479 in piR-598 increases glioma risk by affecting the tumor-suppressive function of piR-598 [
102].
Finally, as we mentioned, miRNAs are crucial key actors in the pathogenesis of most human tumors [
103]. Therefore, because circRNAs function as miRNA modulators, these in turn are also linked to cancer. Currently, a small number of circRNAs have been discovered with several targets’ sites for a particular miRNA, but it has also been observed that most of the circRNAs have, in fact, other functions besides the regulation of miRNA [
104]. For example, it has been revealed that circRNAs are highly expressed in several tumor cell lines from the ENCODE consortium data [
105]. These studies have demonstrated the involvement of circRNAs in cancer through unexpected functions. This is the case of colorectal carcinoma (RCC) in which Bachmayr-Heyda et al. confirmed a general decrease in circRNAs expression levels in tumor tissue compared to adjacent non-cancerous tissues [
106]. This reduction has been negatively correlated with proliferation. The authors affirmed that circRNAs are accumulated in stable-arrested cells, while they are disseminated among the proliferating ones.
2.2. Neurodegenerative Diseases
Although ncRNAs are expressed in all cell types, the Central Nervous System (CNS) is especially enriched [
107]. Thus, approximately 40% of the genes encoding lncRNA are specifically expressed in brain tissue. Other types of non-coding RNAs, such as circRNA and certain miRNAs, are also specifically abundant in the CNS (and some of them specifically involved in synapses) [
108,
109]. miRNAs such as miR-124 and miR-132 have a regulatory impact on neurogenesis [
110], while other lncRNAs, as rhab-domyosarcoma 2 associated transcript (RMST) and Tcl1 upstream neuron-associated long intergenic ncRNA (TUNA), stimulates neuronal differentiation [
111]. In addition, ncRNAs enriched in synaptic connections are known, for example certain miRNA precursors and several miRNAs (e.g., miR-9, miR-132, miR-134, and miR-138) [
112]. Moreover, most circRNAs that are expressed in the brain act as synaptic regulators to regulate local protein expression.
In the case of Parkinson Disease (PD), it has been found that there is a negative correlation between the levels of expression of certain miRNAs and two of the genes involved in this disease: α-synuclein (SNCA) and leucine-rich repeat kinase2 (LRRK2). For instance, miR-7 and miR-153 [
113] are natural regulators of SNCA and LRRK2. Both miRNAs are highly expressed in the brain [
114]. In addition, a specific circRNA related to this disease has also been found through the inhibition of miR-7. Recent research has revealed that circRNA CDR1 acts as a negative regulator of miR-7 causing increased expression of SNCA, which is involved in the development of PD and contributes to oxidative stress. Therefore, circRNA CDR1AS as a miR-7 sponge plays a crucial role in PD by repressing miR-7 [
61].
GWAS studies in PD cohorts have revealed that polymorphisms such as rs2070535 and rs10849446 are located in genes highly related to PD, as PDCK and SCNN1A [
115,
116]. Further studies on these polymorphisms have determined that rs2070535 is located in the 3′-UTR region of PDCK, and differential expression of miRNAs may influence the changes in expression of this gene as seen in this disease. Similarly, rs10849446 is located in a intronic region of SCNN1A related to lncRNAs [
117].
LncRNAs involved in PD have also been discovered, for example the antisense transcript of the UCHL1 gene (UCHL1-AS) is also linked to the prognosis of this disease. The Ubiquitin carboxyl-terminal hydrolase isozyme L1 (UCHL1] is highly expressed in the substantia nigra and related to neuron differentiation. Several authors had pointed out that UCHL1 is a PD risk gene [
118]. In that sense, UCHL1-AS targets Uchl1 mRNA to induce its translation and increasing UCHL1 levels [
43].
Alzheimer’s disease (AD) is another type of neurodegenerative disorder caused by many different factors. Among them, we can highlight the following ones related to ncRNAs. Elevated levels of amyloid-β protein (Aβ), BACE1 proteins, as well as the NAT generated in the strand opposite to this gene, BACE1-AS, have been detected in AD. Aβ is resulting from the cleavage of the amyloid precursor protein (APP) by the APP enzyme of the beta 1 site (BACE1) and the γ-secretase complex. The aberrant expression of these molecules has been related with numerous neurological syndromes and mainly in subjects with Alzheimer’s disease, which emphasizes the importance of regulating the catalytic activity of BACE1 in this disease. In that sense, it was discovered that the CIRS-7 circRNA, downregulated in AD brain tissue, has the main function of reducing APP and BACE1 protein levels [
119]. In addition, other set of circRNAs has been implicated in AD through the alteration of myelin function [
120].
Another example is 17A, this is a ncRNA of a size of 159 nt from the third intron of the G 51 gene receptor coupled to the G protein (GPR51). This ncRNA is involved in increasing expression of the GABA B receptor affecting all signaling cascades dependent on GABA B. ncRNA 17A is overexpressed in AD patients versus healthy tissues, proposing that it might be involved somehow in the initiation of AD [
121,
122].
Furthermore, miRNAs are also associated in the deposition of Aβ, the formation of neurofibrillar nodes (NFT) and extensive neuronal degeneration in the brain. It has been found that miRNAs are able to control APP expression in several ways: for example, miR-106a, miR-520c, members of the miR-20a family (miR-20a, miR-17), miR-16, miR-101, miR-147, miR-655, miR-323-3p, miR-644, and miR-153 are able to bind to a specific sequence in the 3′UTR of APP. Through experimental in vitro studies, the reduction in the expression level of APP when co-expressed with these miRNAs has been verified [
123,
124].
Single nucleotide polymorphisms in ncRNAs have been also associated with AD. It is important to mention that rs7232 and rs12453 in lncRNA NONHSAT160355.1 significantly downregulate the expression of a known AD pathogenic gene, TCN1 in Temporal Cortex, which participates in the regulation of homocysteine in brain to increase risk of AD [
125].
2.2.1. Cardiovascular Diseases
Cardiovascular diseases (CVD) are considered the first cause of mortality all over the world. This concept involves not only heart diseases but also blood vessel associated diseases. The main risk factors classically associated with these diseases are hypertension, diabetes, and smoking, and can be used for stratification and prediction factors for prognosis of the patients. The rest of the risk factors can vary depending on the pathology examined.
Due to the high number of diseases associated to this term, in this section we are going to examine the main ncRNAs historically associated with the development of CVD, and two different cardiovascular pathologies such as atherosclerosis and myocardial infarction.
The lncRNA Braveheart (lncRNA-Bvhrt) was the first one described with a functional implication in cardiac function in mouse heart development. This lncRNA has been associated to the cardiac linage establishment during cardiac cells differentiation [
126]. This role was performed through the interactions with other gene implicated in cell differentiation, SUZ12 (part of the polycomb-repressive complex 2, PRC2), pointing out to the possible role of lncRNA-Bvht in the epigenetic regulation of cardiac commitment [
127]. Furthermore, it suggested the implication of lncRNA-Bvht in directing the neonatal Cardiomyocytes (CM) to a cardiac differentiation. It has been described its involvement in mouse stem cells differentiation to CMs. Other lncRNA called lncRNA-fendrr was described as a proper modulator of heart differentiation at chromatin level [
128].
The implication of other ncRNA, lncRNA-HBL1 (Heart Break LncRNA1) was recently unveil in human induced pluripotent stem cells (hiPSCs) acting as a regulator of CM development. The overexpression of this lncRNA inhibit the CM differentiation from hiPSCs. This is not a direct procedure due to the mechanism of sequestration of hsa-miR-1 during the process. The deregulation of the expression of lncRNA-HBL1 has been described in ischemic heart failure and several cardiovascular diseases in mouse models and human [
129]. Moreover, every cardiac pathophysiology has some lncRNAs which are different for the particular cell type related to each condition.
In contrast with other diseases, in cardiovascular diseases several lncRNAs have been described associated to their development and because of their potential use as a biomarker. In recent studies, some specific lncRNAs were associated and experimentally validated with CVDs: lncRNA ANRIL (antisense noncoding RNA located in INK4 locus), MALAT1 (metastasis associated lung adenocarcinomas transcript 1), KCNQ1OT1 (KCNQ1 overlapping transcript 1), aHIF (natural antisense transcript derived from HIF1alpha), and MIAT (myocardial infarction associated transcript). In that sense, the 9p21.3 risk locus, identified in several genome-wide association studies (GWAS) for coronary artery disease (CAD) susceptibility, is adjacent to the last exons of ANRIL which encompasses multiple SNPs [
130].
The potential therapeutic application of these lncRNAs in atherosclerosis was pointed out since the expression levels of MIAT and ANRIL were higher compared with a non-atherosclerotic tissue control, as well as the expression levels of MALAT were lower than control [
131].
Other classes of ncRNAs, as miRNAs, have been also linked to the development of cardiovascular diseases. Several miRNAs currently related with cardiovascular disorders were previously described as onco-miRs, in association with different cancer types, or related with other cellular functions that are not directly associated with CADs. Some miRNAs like miR-126-3p or miR-21-5p play a key role in cardiovascular diseases such as the response to ischemia process, regulating myocardial fibrosis, ventricular remodeling, arrythmia, or heart failure. In a similar manner, rs687289-A and rs532436-A are intronic variants to the ABO gene relevant to thrombosis and atherosclerosis risk. Nikpay M. et al. stated that rs532436-A and rs687289-A are associated to an increase level of miR-10b-5p, and higher circulating level of miR-10b-5p is associated with increased risk of CAD [
132].
Recently, it was published the updated version of HMDD2.0 database [
133]. This database included experimentally validated associations between miRNAs and different diseases, and included 165 cardiovascular-related miRNAs, spread across the human genome.
Within the definition of Cardiovascular diseases, atherosclerosis is one of the main concerns in the field. It has been defined as a multifactorial disease that involves multiple associated mechanisms, traditionally described as a chronic inflammatory and lipid disorder, and implicating diverse cell types [
134]. During the study of atherosclerosis and its vascular-associated complications, different evidences have shown the involvement of ncRNAs in the development of this disease, and it is considered as other epigenetic mechanism implicated. Even more, the epigenome-wide associated studies allowed to observe the underlying implication of the epigenetic regulation of several related functions with the development of atherosclerosis, such as inflammation, lipid metabolism, and redox cellular status.
Maegdefesset and colleagues were investigating the advantages of a local delivery of some mimic miRs to increase the expression of miR-21 and miR-210 [
135,
136]. Replacing the lack of these miRNAs, conferred stability to the atherosclerotic plaque, decreasing the risk of atherothrombotic vascular complications. Recent studies found a useful signature of five plasmatic miRNAs (circulating miRNAs) for predicting Myocardial Infarction (MI) with a percentage of accuracy of the 81.8% in women and the 74.1% in men. This panel of circulating miRNAs was generated using the data extracted from the HUNT study [
137] and included the following miRNAs: let-7g-5p, miR-106a-5p, miR-424-5p, miR-144-3p, and miR-660-5p. Other circulating miRNAs, which expression is p53-dependent, were described as a novel predictor of heart failure after suffered from MI: miR-192-5p, miR-194-5p, and miR-34a-5p [
138]. General speaking, there is more than 60 miRNAs already associated with CVDs that could be used with diagnostic and prognostic purposes.
The association of another circulating lncRNAs, Zinc finger antisense 1 (ZFAS1) and CDR1 antisense (CDR1AS), has been described as differentially expressed in acute MI patients in comparison with healthy individuals [
139]. The urothelial carcinoma-associated 1 (UCA1) is a lncRNA that was previously associated to bladder and lung cancer, and it was known as a predictive biomarker for its expression in this kind of cancers. Furthermore, it was described its expression in heart tissue of healthy individuals. Recent findings have shown its deregulation from early stage of acute MI until three days after the MI. Moreover, there is an inverse correlating among circulating UCA1 levels and miR-1 expression levels [
140].
The presence of circRNAs in human heart tissue is directly related with the abundance of their related mRNAs. Some of the most important cardiac expressed transcript, as RYR2, DMD, and TTN genes, generate the most abundant circRNAs in heart tissue. In atherosclerosis, CDKN2B-AS1 is perhaps one of the molecularly best-studied circRNAs. According to other studies, this circular RNA from the 9p21 locus, contains a vast number of single nucleotide polymorphisms that have been linked to atherosclerotic vascular disease, as well as to type 2 diabetes mellitus (T2DM) [
141].
2.2.2. Other Diseases
The purpose of this section is to include some of the remaining diseases that could obtain a benefit from the research in ncRNAs as a potential therapeutic target, as well as biomarkers for their use in diagnosis and prognosis of the different pathologies.
Type 1 diabetes is a chronic disease in which very low or absent insulin production by the pancreas has two main consequences: insulitis and pancreatic beta cells destruction. In the organism response to this kind of stress, the pro-inflammatory cytokines interferon gamma (IFNG) and interleukin 1 beta (IL1B) are produced during insulitis and enhance endoplasmic reticulum (ER) stress response and trigger the expression of BCL2-related proteins in beta cells, participating in their cell death. Grieco et al., has recently unveil the implication of the downregulation of two miRNAs associated to these facts from the same miRNA family, miR-204-5p, and miR-211-5p [
142]. These miRNAs have been involved in the regulation of the transcriptional expression of some ER stress genes downstream PERK, specifically DDIT3 (CHOP, pro-apoptotic protein). These novel findings related with early type 1 diabetes point out a relationship between miRNAs, ER stress and beta cell apoptosis.
Type 2 diabetes is a form of the disease characterized by three different conditions in the human body: high levels of glucose in blood, a decrease in the amount of insulin, and an insulin resistance. This is another kind of defect of the pancreas and it is caused by an impairment between the insulin sensitivity and the insulin secretion.
The involvement of miRNAs in the production of insulin has been extensively studied. miRNAs play an important role in the regulation of the gene coding for the insulin. The expression of several miRNAs has been associated with this process. For instance, the upregulation of miR-30d directly correlated with high glucose levels. This fact has been previously described by Tang et al., enhancing the transcription of the insulin gene, and the opposite effect was observed during its inhibition [
143]. Another important miRNA in the pancreas is miR-375. Its overexpression is able to inhibit the glucose-stimulate insulin expression. This miRNA achieves this effect by means of the regulation of 3′-phosphoinositide-dependent protein kinase-1 (PDK1), unveiling its important role within the pancreas [
144,
145,
146].
The main organs depending on the glucose consumption in the organism are the liver and the skeletal muscle. Recent studies have also described the implication of other miRNAs in mechanisms associated to insulin resistance. For example, miR-29 has been described by Zhou et al., in the insulin resistance by targeting PPARδ in skeletal muscle [
147]. In mice, the upregulation of miR-29 was also related with the inhibition of the gluconeogenesis. In mice liver, miR-33 regulates the expression of insulin receptor substrate 2 (IRS2), that is related with insulin resistance [
148].
There are many renal diseases associated with the dysregulation of ncRNAs, specifically miRNAs, such as acute renal damage, renal cell carcinoma, diabetic nephropathy, polycystic kidney disease (PKD), and others. In the development of these diseases, it has been described an important involvement of miRNAs. Different studies were coincident in the importance of miRNAs and their use as biomarkers for the diagnosis of these renal diseases. The miR-200 family has been described as abundant in renal tissue and highly expressed [
149]. Some of the components of this family has been found in a very low expression in patients suffering from IgA neuropathy, being the level of decrease of the expression considered as a prognostic factor in this disease. Moreover, the increase on the expression levels of miR-155 and miR-146a has been also correlated with a poor prognosis of the disease, at the clinical and pathophysiology level [
150].
The deregulation of the expression of miRNAs can be used as a diagnostic or prognostic factor for several diseases, such as cancer. In this specific case, they could be useful in the detection of diseases related with the presence of cysts, like PKD, or associated with a process of cystogenesis in the organs with like liver, pancreas, and ovary. Thus, these miRNAs could be used as a novel biomarker for detecting renal diseases.
In the case of autoimmune diseases, there are also ncRNAs implicated at different levels. As an example, rs57095329 is located in the miR-146a promoter which confers risk of systemic lupus erythematosus (SLE). It turns out that individuals carrying the risk allele in the promoter showed lower expression levels of miR-146a [
151]. Further, in SLE, the over expression of linc00513 plays a role in lupus pathogenesis by promoting IFN signalling pathway. SNP variants in the linc00513 promoter are functionally significant in regulating linc00513 expression and conferring predisposition to SLE [
152].