1. Introduction
The single-layer epithelium lining the intestinal tract is integral to its functions in water and nutrient absorption, waste elimination, and immune surveillance while also forming a barrier against luminal toxins and gut-resident microbiota. To weather the barrage of chemical, pathogenic, and mechanical stresses posed by the digestive process, and counterbalance cell attrition, the intestine must continuously replenish its epithelial lining and regenerate the full gamut of specialized cell types that underpin its diverse functions. The homeostatic renewal of this epithelium is critically dependent on the sustained activity of multipotent intestinal stem cells (ISCs), residing within submucosal invaginations, called crypts. ISCs can self-renew while also giving rise to short-lived transit-amplifying (TA) cells, which in turn undergo successive rounds of cell division to generate multiple mature intestinal cell types (). Broadly, differentiated intestinal cell lineages are specialized to perform either absorptive or secretory functions [
1]. Absorptive enterocytes retrieve nutrients and water from luminal contents, whereas rare microfold (M) cells function in immune surveillance, delivering luminal antigens to underlying lymphoid structures (Peyer’s patches). Secretory lineages include multiple hormone- and neurotransmitter-secreting enteroendocrine cell types that regulate physiological responses to food intake and interface with the enteric nervous system, mucus-secreting goblet cells that fortify the host epithelium against mechanical stresses and luminal microorganisms, and chemosensory tuft cells that orchestrate type-2 immunity responses to helminth parasites and allergens [
1].
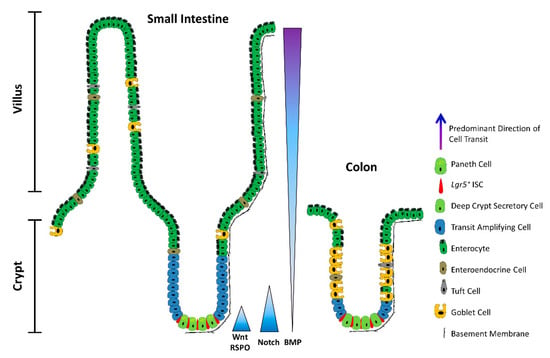
Figure 1. The architecture of the small intestine and the colon. Schematic depicting a longitudinal section of the intestinal mucosa. The mucosa of the small intestine extends finger-like projections (villi) into the gut lumen, which provide an increased surface area for optimal nutrient absorption. The villi are populated by mature, differentiated absorptive and secretory cell types, including absorptive enterocytes, hormone- and neurotransmitter-secreting enteroendocrine cells, mucus-secreting goblet cells, tuft cells, and microfold (M) cells (not shown). The mucosa surrounding the villi forms tubular invaginations into the lamina propria, called crypts, which serve as a protected reservoir of stem and progenitor cell populations. Notably, the epithelium of the colon is devoid of villi, with the crypts opening onto a flat mucosal surface, reflecting its role in waste compaction. To support homeostatic turnover, ISCs self-renew and give rise to short-lived transit-amplifying (TA) cells, which in turn beget lineage-restricted progenitors that differentiate into the mature cell types lining the villi. During their limited lifespan, intestinal epithelial cells migrate from the base of the crypt to the tip of the villus or the colonic surface, from where they are shed into the gut lumen and replaced by neighbouring cells. In contrast, Paneth cells are relatively long-lived, and migrate to the base of the crypt, where they secrete antimicrobial peptides and form a vital component of the ISC niche. Paneth cells are absent from the colon, but deep crypt secretory (DCS) cells may fulfil an equivalent role. Opposing gradients of morphogens specify intestinal-cell fate and differentiation along the vertical crypt axis: Wnt and Notch signalling prevail at the crypt base, whereas BMP transduction is highest near the lumen.
Since aberrant ISC proliferation or, conversely, the failure to mobilize ISCs in response to injury is invariably detrimental, ISC activity is kept in check by the local milieu: the ISC niche. Set within the confines of the crypt base, the ISC niche is comprised of either Paneth cells in the small intestine or deep crypt secretory (DCS) cells in the colon, in addition to pericryptal fibroblasts, immune cells, endothelial cells, enteric neurocytes, extracellular matrix (ECM) components, and soluble cytokines and growth factors. Multiple converging niche-signalling pathways—primarily Wnt, Notch, and BMP—maintain ISCs in a multipotent state and fine-tune the balance between self-renewal and differentiation.
2. ISCs in a Nutshell
Daily homeostatic turnover of the intestinal epithelium is orchestrated by crypt-base columnar cells, nestled between either Paneth or DCS cells at the crypt base in the small intestine and colon, respectively [
2,
3]. Decorated with the RSPO-receptor LGR5, which potentiates canonical Wnt/β-catenin signalling [
4,
5,
6], these highly proliferative cells (hereafter
Lgr5+ ISCs) exhibit the ability to self-renew and differentiate into all intestinal lineages in vitro and in vivo [
7,
8] and are tasked with the homeostatic renewal of the epithelium in both the small intestine and the colon [
7] (a). Yet, remarkably, the adult intestinal epithelium can fully recover following acute ablation of
Lgr5-expressing cells [
9], and conditional deletion of the
Lgr5 gene does not visibly perturb crypt architecture [
4]. Together, these findings bring forth the redundancy of
Lgr5+ ISCs for homeostasis and suggest that other cell types can compensate for their deficiency in this setting. Nevertheless, sustained depletion of
Lgr5+ ISCs severely compromises the regenerative response to radiation-induced damage [
10], suggesting that any compensatory cell types, deployed post injury, must first repopulate the
Lgr5+ compartment prior to reconstituting the lost epithelium. Indeed, multiple putative reserve ISC pools have been proposed to reside just above the crypt base—around the so-called +4 position—based on DNA-label retention [
11] and lineage tracing with eGFP−IRES−Cre
ER reporters inserted into the loci encoding BMI1 [
12], mTERT [
13], HOPX [
14], or LRIG1 [
15]. These slow-cycling reserve populations can be mobilized post injury to replenish lost or damaged
Lgr5+ ISCs [
9,
12,
13,
14,
15,
16,
17,
18,
19]. In addition, multiple lineage-committed progenitors and fully differentiated cell types can dedifferentiate and regain stem-like traits. Notably,
Alpi+ enterocyte precursors [
20],
Prox1+ enteroendocrine-lineage cells co-expressing tuft-cell markers [
21] and other subsets of enteroendocrine cells [
22,
23,
24], CD69
+CD274
+ goblet cell precursors [
25], secretory progenitors expressing
Dll1 [
26] or
Atoh1 (also known as
Math1) [
27,
28,
29,
30], differentiated KRT20
+ surface enterocytes in the colon [
31], as well as post-mitotic tuft [
32], enterochromaffin [
33], and Paneth cells [
34,
35,
36] can contribute to varying degrees to crypt homeostasis and injury-induced regeneration (b).
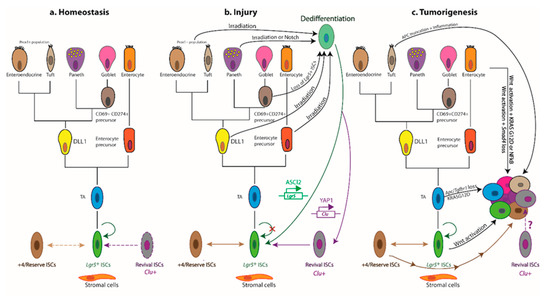
Figure 2. Cellular hierarchies and phenotypic plasticity in the intestinal epithelium during homeostasis, post-injury regeneration, and tumorigenesis. The dendrograms summarize key lineage relationships between Lgr5+ ISCs, transit-amplifying (TA) cells, lineage-committed precursors, terminally differentiated intestinal cell types, +4/reserve ISCs, and revival stem cells in different settings. (a) The homeostatic remodelling of the intestinal epithelium is orchestrated by niche signals that fine-tune the balance between Lgr5+ ISC self-renewal and differentiation. Revival stem cells are rare under homeostatic conditions (dashed boundary), and +4/reserve ISCs contribute only weakly to daily turnover during homeostasis (double-headed dashed arrow). (b) Following ablation of Lgr5+ ISCs post injury, +4/reserve ISCs can mobilize to replenish lost Lgr5+ ISCs and repopulate damaged crypts. Differentiated Lgr5+ progeny upregulate Clu in a YAP1-dependent manner, transiently serving as revival stem cells that can generate Lgr5+ ISCs de novo. Lineage-committed precursors and/or fully differentiated cells can dedifferentiate, revert to an Lgr5+ state, and regain stem-like traits. The transcription factor ASCL2 is critical for the ability of recent Lgr5+ ISC progeny to undergo dedifferentiation to an Lgr5+ state. Of note, Clu+ cells are distinct from the Lgr5+ and Ascl2+ populations. (c) Aberrant activation of Wnt signalling drives unbridled proliferation of Lgr5+ ISCs leading to intestinal hyperplasia. TA and differentiated cells, with hyperactive Wnt signalling and mutant KRASG12D, may progress to malignancy in the context of TGFβ-receptor loss or inflammation (i.e., NFκB activation). Multiple +4/reserve ISCs can also initiate tumorigenesis, and tuft cells are readily transformed by inflammation following Apc loss. Whether revival stem cells can serve as tumour-initiating cells remains unclear. Solid arrows indicate the ability to dedifferentiate and revert to a stem-like state, or the susceptibility to transformation and hyperplastic progression. Reflexive arrows indicate the ability to self-renew. Double-headed solid arrows denote dynamic interconversion between indicated cell types. Note that, to date, goblet cell progenitors have not been lineage-traced.
Classification of ISCs is further confounded by the expression of markers of +4/reserve ISCs (
Bmi1,
mTert,
Hopx,
Lrig1) in
Lgr5+ cells, suggesting considerable overlap between these populations [
37,
38,
39]. Indeed,
Lgr5+ and +4/reserve ISCs can dynamically interconvert during both homeostatic and radiation-induced regeneration, although +4/reserve ISCs contribute only weakly to daily turnover under non-pathological conditions [
9,
14,
16,
17]. Nevertheless, most +4/reserve ISC populations (including
Dll+ secretory precursors) differ profoundly from
Lgr5+ ISCs as they are relatively refractory to Wnt stimulation, lack expression of canonical Wnt-target genes, and exhibit resistance to high-dose irradiation [
14,
16,
17,
40,
41].
In addition to
Lgr5—itself a Wnt-target gene—
Lgr5+ ISCs express other Wnt targets, including
Ascl2,
Sox9,
Troy, and
Axin2 [
39,
71,
72], underscoring the importance of Wnt signalling for safeguarding the stem cell state at the crypt base [
72]. ASCL2—a master regulator of the
Lgr5+ ISC gene expression program—perpetuates its own expression in a positive feedback loop, controlled by WNT/RSPO levels. Thus, while an “
Ascl2-on” state imparts stemness, “
Ascl2-off” cells are destined to differentiate, with the corollary that TA cells can regain stemness upon encountering increased WNT/RSPO levels sufficient to drive
Ascl2 expression [
73].
Recent insights have further highlighted heterogeneity within the
Lgr5+ compartment itself. Addressing a long-standing controversy in the field, Buczacki and colleagues identified label-retaining cells as
Lgr5+eGFP
hi secretory precursors of Paneth and enteroendocrine cells that do not contribute to homeostasis [
30] and are discrete from the +4/reserve ISCs marked by Cre
ER knock-in reporters [
42]. Two additional slow-cycling
Lgr5+ ISC subpopulations, expressing
Mex3a [
43] or
Krt15 [
44], were found to survive genotoxic stress and contribute to radiation-induced regeneration. In this respect, these slow-cycling
Lgr5+ ISC subsets exhibit purported traits of +4/reserve ISCs and markedly contrast with the highly proliferative, radio-sensitive
Lgr5+ ISC population. Furthermore, a rapidly-cycling, DNA damage-resistant subpopulation of
Msi1+ cells, that expresses little-to-no
Lgr5 and resides at the +4 position, has recently been shown to repopulate the intestinal epithelium post irradiation [
45]. Crucially,
Msi1+ cells are mobilized before the reappearance of
Lgr5+ cells, challenging the widely held contention that +4/reserve ISCs must regain
Lgr5 expression prior to instigating repair [
46]. Although able to repopulate all major intestinal lineages,
Msi1+ cells preferentially differentiate into Paneth cells, suggesting that they may first replenish the ISC niche to help restore
Lgr5+ ISC functionality in the newly remodelled crypt [
45]. An additional distinct—but transient—population comprises the immediate progeny of
Lgr5+ ISCs, expressing modestly reduced levels of ISC-associated transcripts alongside markers of mature secretory cells and enterocytes [
47]. An example of multilineage gene priming, this transient bipotential progenitor population is poised to lose
Lgr5 expression entirely as cells move further from the crypt base along their ultimate cell-fate trajectory [
47,
48]. Collectively, these findings suggest considerable overlap and dynamic interconversions between crypt ISC populations and implicate the local niche as the main “influencer” of stem-like behavioural and phenotypic traits.
Bringing a long-standing debate to an apparent close [
49], recent studies have attributed the bulk of intestinal epithelial regeneration to the dedifferentiation of recent progeny of
Lgr5+ ISCs. Both absorptive and secretory cell lineages are recruited to replenish the stem-cell pool post injury, with the underlying kinetics precluding the mobilization and expansion of dedicated reserve ISC populations [
50]. In fact, the induction of
Ascl2 is critical for the ability of recent
Lgr5+ ISC progeny to undergo dedifferentiation to an
Lgr5+ state prior to regenerating the injured intestinal epithelium [
50]. Mechanistically, such pervasive dedifferentiation is underpinned by a permissive open chromatin configuration in progenitor cells undergoing differentiation [
51], with only incremental chromatin remodelling of lineage-restricted genes required to interconvert between homeostatic
Lgr5+ ISCs and their secretory and absorptive eventual progeny during differentiation, and vice versa during crypt regeneration [
25,
51].
While at times confounding, these studies collectively converge on the fact that most, if not all, crypt-resident cell types display phenomenal plasticity and retain (dormant) stemness potential, calling into question the existence of a “dedicated” reserve ISC pool. Importantly, they underscore the notion that stemness is not a cell-intrinsic trait and refocus attention on the role of the niche in governing ISC function during homeostasis and the de novo acquisition of stemness in times of stress.
3. The YAP-Driven Foetal-Like Stem Cell State
Additional studies have interrogated the molecular mechanisms underlying the response of the mouse intestinal epithelium to helminth infection [
148] and treatment with dextran sodium sulphate (DSS) [
149], both of which breach the mucosal barrier. An emergent theme is that the regenerating intestinal epithelium is transiently reprogrammed into a highly plastic foetal-like state, orchestrated by changes in the inflammatory milieu [
148] and ECM [
149], respectively. The extensive tissue remodelling that ensues entails the deployment of highly proliferative SCA1
+ progenitors [
148,
149], lacking markers of secretory lineages as well as of adult ISCs—most notably,
Lgr5 and LRIG1 [
39,
149]. Instead, these regenerating cells express foetal epithelial markers, such as
Anxa1 and
Tacstd2/
Trop2, alongside the multipotent progenitor marker SCA1 (also known as LY6A) [
149,
150]. Notably, although
Sca1 is absent from the human genome,
ANXA1 is highly expressed in the regenerating epithelium of inflamed ulcerative colitis, compared with non-inflamed regions in matched patient specimens [
149]. Moreover, the transcriptional signatures of the mouse repairing epithelium and the foetal-like state are enriched in patients with active inflammation, compared with healthy counterparts, lending relevance of this foetal-like program to human disease [
149]. Similarly, crypts overlying helminth larvae-associated granulomas become devoid of
Lgr5 expression, with a discrete subset of SCA1
+ crypt cells activating an IFNγ-dependent foetal-like transcriptional program [
148]. Indeed, similar injury-response programs are deployed following irradiation and
Lgr5+ ISC ablation, suggesting that the transient “revival” of latent foetal-like traits is likely a common denominator of the intestinal epithelial response regardless of the mode of injury [
148].
Following DSS treatment, the regenerating mouse intestinal epithelium is also characterized by upregulation of several ECM components and the accumulation of collagen type I fibres around newly formed crypts. These dynamic changes in the ECM composition of the niche are propagated via FAK/SRC-mediated mechanotransduction, culminating in the activation and nuclear translocation of YAP and TAZ [
149], two paralogous transcriptional coactivators inhibited by the Hippo tumour-suppressor pathway [
151]. YAP has similarly been shown to transiently reprogram
Lgr5+ ISCs into a regenerative state post irradiation. Here, YAP suppresses homeostatic Wnt signalling and Paneth cell differentiation while concomitantly activating expression of the EGF-family member EREG to drive proliferation and promote cell survival [
152]. Indeed, several studies concur that YAP/TAZ can inhibit Wnt signalling during intestinal regeneration and tumorigenesis [
152,
153,
154,
155], consistent with the suppression of
Lgr5 and the ISC signature during the foetal-like regenerative response [
149].
A critical role for YAP has also been ascribed in the damage-induced mobilization of “revival stem cells”, recently identified in the regenerating intestinal epithelium using a single-cell transcriptomics approach [
156]. The revival stem cell pool is a rare, quiescent population in homeostasis, characterized by elevated expression of clusterin (
Clu),
Anxa1,
Cxadr, and
Basp1. While these
Clu+ revival stem cells do not contribute to daily homeostatic renewal, they are mobilized and expanded following ablation of
Lgr5+ ISCs, irradiation, or DSS-induced inflammation and colitis. Their transient expansion post damage regenerates the full gamut of intestinal cell types, including
Lgr5+ ISCs, in a YAP1-dependent manner [
156]. Interestingly,
Clu+ revival stem cells express elevated levels of
Sca1 post irradiation [
156], raising the possibility that this damage-induced, expanded revival stem cell population overlaps with
Sca1+ foetal-like crypt cells, which also rely on YAP for their regenerative potential [
149].
Whereas the YAP-driven regenerative response is a transient, reversible process [
149,
156], persistent tissue injury and repair set up a vicious cycle of chronic inflammation—a known risk factor for colorectal cancer (CRC) [
143]. Indeed, the YAP-mediated regenerative response can be hijacked to facilitate the progression of APC-deficient foci to adenomas to the extent that
Yap deletion abrogates adenoma formation in
ApcMin/+ mice [
152,
158]. Moreover, the YAP transcriptional program correlates with the gene expression signatures of early
ApcMin/+ tumours as well as of revival stem cells [
156,
159]. Accordingly, YAP decorates the nuclei of tubular adenomas from patients afflicted with familial adenomatous polyposis [
158]. Yet, the role of YAP in intestinal tumorigenesis remains controversial as both tumour-suppressive [
153,
155] and oncogenic functions [
152,
158,
160,
161] have been ascribed in different contexts. It further remains to be seen whether the foetal-like, YAP/TAZ-dependent,
Lgr5− regenerative state plays a role in the development of colonic tumours lacking overt Wnt-pathway mutations. In support of this notion, BRAF
V600E-driven colonic organoids exhibit a foetal-like dedifferentiation program enriched for Hippo-pathway targets, which recapitulates the transcriptional profiles of human BRAF
V600E-driven CRCs [
162]. In addition, the acquisition of a YAP/TAZ-dependent foetal-like signature is associated with resistance to Wnt-targeted therapy [
163], and
Sca1+ reserve-like stem cells, with a regenerative/tumorigenic YAP transcriptional signature and concomitant suppression of β-catenin signalling, fuel adenoma initiation in
ApcMin/+ mice as well as an azoxymethane-induced tumour model. Crucially, the tumorigenic capacity of these
Sca1+ reserve-like stem cells depends on the druggable PGE2–PTGER4 axis, which in turn controls the nuclear localization/activity of YAP [
159]. Indeed, PGE2-induced YAP signalling is also implicated in colitis-associated regeneration and spontaneous tumorigenesis [
164], suggesting that the pro-oncogenic, YAP-dependent, foetal-like regenerative program may serve as a therapeutically actionable target.
Overall, multiple cell types can mobilize to regenerate the injured intestinal epithelium by adopting a highly plastic foetal-like state, although the degree to which each population contributes to the repair warrants further study. It also remains unclear whether regenerative cues can mobilize different crypt progenitors and/or mature cell types to dedifferentiate into a foetal-like state, and/or whether pre-existing homeostatic crypt cell types, such as the
Lgr5−Clu+Sca1+ revival stem cells [
156], expand in an attempt to restore the epithelium independently of Wnt signalling. Furthermore, whether revival stem cells can serve as tumour-initiating cells remains untested at present. For example, it is conceivable that PGE2-dependent
Sca1+ reserve-like tumour-initiating cells [
159] derive from transformation of
Lgr5−Clu+Sca1+ revival stem cells [
156]. Consistent with this notion, the revival stem cell signature correlates with resistance to 5-fluorouracil chemotherapy in patient-derived CRC organoids, and elevated
CLU expression is associated with poor patient survival and disease recurrence [
165]. In addition, the revival stem cell signature is reportedly enriched in L1CAM-positive metastasis-initiating CRC cells [
166].
4. Cells-of-Origin and Plasticity in Colorectal Cancer
Analogous to normal crypts, current dogma posits that only a subset of intestinal tumour cells—termed cancer stem cells (CSCs)—are endowed with tumour-initiating potential, i.e., the capacity to self-renew and generate the differentiated non-CSCs that constitute the tumour bulk. CSCs are also thought to underpin metastatic competence, drug resistance, disease recurrence and, ultimately, poor therapeutic outcome. In line with the pervasive plasticity of the intestinal epithelium, accumulating evidence supports both “bottom-up” [
273] and “top-down” [
281] histogenesis of colorectal tumours whereby the cells-of-origin comprise either ISCs at the crypt base or differentiated cells at the crypt apex, respectively (c).
Lgr5+ ISCs have been amply demonstrated to serve as tumour-initiating cells [
59,
282]. Indeed, targeted deletion of
Apc in
Lgr5+ ISCs drives aberrant Wnt signalling and hyperproliferation, leading to rapid adenoma formation in mice [
59]. Moreover, overexpression of
Rspo3 in
Lgr5+ cells drives hyperplastic bottom-up lesions, containing mislocalized Paneth cells and expanded
Lgr5+ and
Lgr4+ populations, in keeping with the fact that RSPO3 is a secreted protein that nurtures both
Lgr5+ ISCs and their supportive epithelial niche. Besides
Lgr5+ cells, however, lineage tracing implicates
Lgr5− populations as putative cells-of-origin of the resulting hyperplastic adenomas and adenocarcinomas in this model [
282]. Similarly, constitutive activation of Wnt signalling in cells expressing
Bmi1 [
12],
Prom1 [
283], or
Lrig1 [
15,
284] drives bottom-up intestinal neoplasia in mice. Collectively, these findings suggest the existence of multiple possible cells-of-origin within the crypt.
Other crypt cell types can also assume the mantle of tumour-initiating cell, contributing to bottom-up tumorigenesis. Hence, loss of
Apc in
Krt15+ cells—a heterogeneous population, encompassing
Lgr5+ and
Lgr5− cells, spanning the crypt base as well as the TA zone—leads to adenomas that occasionally progress to invasive adenocarcinomas [
44]. Such lesions are not typically observed upon sole deletion of
Apc in other putative tumour-initiating cell populations, including
Lgr5+ ISCs [
15,
18,
59,
284]. It remains to be determined whether the coexistence of adenomas and adenocarcinomas—frequently a feature of human polyposis syndromes—reflects tumour initiation from distinct differentially localized subsets of
Krt15+ cells [
44]. Remarkably, while the majority of
Lgr5+ subsets are exquisitely sensitive to DNA damage,
Krt15+Lgr5+ cells are radioresistant and may thus survive to spawn tumours post injury.
Conversely, top-down lesions likely derive from cells located in the TA zone or the villus, induced to undergo dedifferentiation. Notably, sole deletion of
Apc in TA cells yields only microscopic lesions, which rarely progress to adenoma [
59]. Additional TGFβ dysfunction is not sufficient to drive dedifferentiation in this compartment or the formation of top-down lesions [
285]. However, following exposure to inflammation and/or upon accumulating cooperating mutations, differentiated villus cells can re-express
Lgr5 and ISC markers, and initiate tumours [
286,
287]. Thus, constitutive activation of β-catenin and NFκB signalling [
286] or dual
Apc/
Kras mutations [
287] can drive tumour formation both from crypt ISCs and villus epithelial cells in the small intestine [
286] and colon [
287], respectively. Deletion of
Tgfbr1 further augments the dedifferentiation potential of
VilCreERApcfl/flKrasG12D/+ villus epithelial cells, exacerbating top-down tumorigenesis [
285]. This suggests that, during early tumour progression, the elevated stromal-derived TGFβ levels that prevail further up the crypt–villus axis restrain dedifferentiation, whereas cells in lower regions or the crypt base can escape to form tumours. Consequently, mutations enabling differentiated cells to evade TGFβ-mediated tumour suppression will extend the pool of tumour-initiating cells.
Additional examples whereby dedifferentiation bestows tumorigenic potential have been reported. Long-lived differentiated
Dclk1+ tuft cells, which remain quiescent following
Apc loss, are readily transformed by inflammation, forming poorly differentiated colonic adenocarcinomas [
32]. APC truncation or post-irradiation depletion of
Lgr5+ ISCs induces radioresistant
Krt19+Lgr5− upper-crypt progenitors to dedifferentiate, via an
Lgr5+ state, spawning tumours both in the small intestine and colon [
18].
Bhlha15+ secretory cell precursors are another candidate cell-of-origin located just above the ISC zone. In the small intestine, these cells can dedifferentiate to form tumours with serrated features upon sustained activation of Notch signalling, combined with
Apc loss. In the colon, the counterpart
Bhlha15+ cell population is mobilized upon DSS treatment via the activation of SRC and YAP [
290]. Thus,
Bhlha15+ secretory cell precursors respond differently to tumorigenic insult in distinct niches, although the clinical relevance of these findings remains unclear [
290].
Aberrant expression of the BMP inhibitor
GREM1 in the intestinal epithelium disrupts homeostatic morphogen gradients, prompting the proliferative expansion of
Lgr5− progenitor cells that spur the formation of ectopic crypt foci perpendicular to the villus axis. Cells, within these structures, accumulate multiple somatic mutations, with concomitant suppression of cytostatic and differentiation programs, eventually progressing to polyps that recapitulate features of hereditary mixed polyposis syndrome and traditional serrated adenomas [
94]. These findings further confirm that
Lgr5− cells, outwith the ISC niche, can undergo malignant transformation.
Together, the above findings reinforce the links between deregulated niche signalling, prolonged inflammation, and CRC risk/progression [
291] (). Crucially, they underscore that the initiation of tumours from differentiated cells requires cooperating mutations or exacerbating stimuli, such as an inflammatory drive, alongside Wnt deregulation.
5. All Roads Lead through LGR5
Notwithstanding the existence of multiple putative cells-of-origin within the crypt base or more differentiated luminal regions, compelling evidence supports the contention that LGR5 marks a subset of mouse and human intestinal CSCs endowed with tumorigenic potential and multi-lineage differentiation capacity [
59,
292,
293,
294,
295,
296,
297,
298]. Perhaps unsurprisingly, considering the pervasive plasticity of the intestinal epithelium, diphtheria toxin-mediated ablation of cells engineered to express the diphtheria-toxin-receptor, DTR, under the control of the
Lgr5 promoter (
Lgr5DTR), failed to achieve regression of non-metastatic
ApcMin/+KrasLSL-G12D/+Vil1Crep53−/−Lgr5DTR/eGFP subcutaneous organoid allografts. Instead, tumours remained in a state of stasis while
Lgr5− populations mobilized to sustain growth, albeit less efficiently than
Lgr5+ counterparts [
297]. Notably, tumour growth resumed unabated following treatment withdrawal, underpinned by dynamic conversion of
Lgr5− non-CSCs into
Lgr5+ cells. Intriguingly, comparable growth dynamics were observed in cultured organoids, suggesting that the repopulation of
Lgr5+ cells may partly rely on intrinsic
Lgr5− cell properties and proceed independently of tumour-activated stroma [
297]. The mechanisms whereby non-CSCs, or distinct subsets thereof, sense the depletion of
Lgr5+ CSCs within a tumour, and the intrinsic and extrinsic cues that trigger their mobilization remain an important avenue for investigation to better understand therapy resistance and tumour recurrence.
Similarly, xenografted patient-derived organoids contain differentiated
KRT20+ cells that can re-express
LGR5 and fuel tumour regrowth [
299]. In this model, short-term ablation of
LGR5+ cells, in combination with anti-EGFR therapy, elicited a more pronounced inhibition of tumour growth than either treatment alone [
299]. Consistent with this, residual drug-resistant
LGR5− cells that can reconstitute tumour growth, following
LGR5+ cell depletion, express the EGF-family member EREG [
300]. Interestingly, oxaliplatin did not synergize with anti-EGFR, owing to the failure of chemotherapy to induce
LGR5 expression in
LGR5− cells [
299]. Since
LGR5+ and
KRT20+ cells appear to reside within distinct tumour niches [
298], it is plausible that depletion of the
LGR5+ population exposes differentiated
KRT20+ cells to aberrant instructive signals that incite their dedifferentiation and acquisition of CSC traits, analogous to the reversion of multiple intestinal cell types to an
Lgr5+ state during injury-induced regeneration [
9,
20,
26].
Until recently, little was known about the identity of metastasis-initiating cells in CRC and their relationship to primary tumour CSCs. Ablation of
Lgr5DTR CSCs in orthotopically implanted
ApcMin/+KrasLSL-G12D/+Vil1Crep53−/−Smad4−/−Lgr5DTR/eGFP organoids demonstrated an indispensable role for
Lgr5+ CSCs in the formation and maintenance of metastatic outgrowths, even though their ablation proved inefficacious in the primary tumour setting [
297]. Most notably, treatment cessation was not accompanied by regrowth of liver metastases, highlighting the potential therapeutic benefits of targeting
Lgr5+ CSCs in the metastatic setting [
297]—the ultimate cause of patient demise. In addition, these findings suggest that distinct tumour cell subsets may harbour differential abilities to drive primary tumour growth and initiate metastases, and underscore the importance of a permissive microenvironment as a prelude for colonization at the distant site [
297].
Unexpectedly, ablation of
Lgr5DTR CSCs did not impair primary tumour invasiveness per se, yet still reduced liver metastatic burden, raising the possibility of LGR5-independent mechanisms of productive invasion [
297]. Indeed, using intravital multiphoton microscopy to observe spontaneous metastatic progression from orthotopically implanted, genome-edited CRC organoids [
301], van Rheenen and colleagues made the striking observation that the majority of circulating tumour cells lacks
Lgr5. In vitro,
Lgr5− cells were intrinsically competent to form organoids and spawn functional
Lgr5+ progeny, independently of niche signals, although the emergence of
Lgr5+ cells was increased in the presence of HGF and FGF [
302]. Importantly, targeted ablation of
Lgr5DTR/eGFP cells prevented the progression of micrometastases, similar to the earlier findings of de Sousa e Melo et al. [
297], with colonization and outgrowth of seeded
Lgr5− cells dependent on the de novo expression of
Lgr5 [
302]. While
Lgr5+ CSCs were detected in the migratory population, they were not typically recovered from the circulation, raising the intriguing possibility that, upon escaping the confines of the primary tumour niche,
Lgr5+ cells enter an
Lgr5− non-CSC state that likely confers the ability to navigate and survive the perils of the metastatic cascade. Following seeding of
Lgr5− cells at the distant site, their reversion to an
Lgr5+ state allows the outgrowth and progression of micrometastases. Although organoid cultures suggest that
Lgr5− non-CSCs can spontaneously revert to an
Lgr5+ state in a niche-independent manner, signals emanating from the tumour microenvironment may nevertheless influence this transition in vivo. Deciphering the signals that instruct the plasticity transitions between
Lgr5+ and
Lgr5− states, and the underlying molecular mechanisms, may yield important insights into critical determinants of disease progression and therapy resistance, and inform new strategies to target metastatic plasticity.
Overall, the above findings attest to the plasticity of Lgr5− tumour-bulk cells, suggesting it may underpin failed treatment outcomes and metastatic competence. Which differentiated/non-CSC Lgr5− subsets are mobilized to replenish primary tumour growth, when the Lgr5+ CSC pool is compromised, and whether these are the same cell subsets that exhibit metastatic competence remains to be seen. The ability of differentiated/non-CSC Lgr5− cells to activate a dormant plasticity program at the distant site, seed metastases, and re-establish a cellular hierarchy de novo highlights the need to target intrinsic plasticity mechanisms as well as extrinsic niche pathways in order to ablate metastatic potential.