Ibrexafungerp (formerly SCY-078 or MK-3118) is a first-in-class triterpenoid antifungal or “fungerp” that inhibits biosynthesis of β-(1,3)-D-glucan in the fungal cell wall, a mechanism of action similar to that of echinocandins. Distinguishing characteristics of ibrexafungerp include oral bioavailability, a favourable safety profile, few drug-drug interactions, good tissue penetration, increased activity at low pH and activity against multi-drug resistant isolates including C. auris and C. glabrata. In vitro data has demonstrated broad and potent activity against Candida and Aspergillus species. Importantly, ibrexafungerp also has potent activity against azole-resistant isolates, including biofilm-forming Candida spp., and echinocandin-resistant isolates. It also has activity against the asci form of Pneumocystis spp., and other pathogenic fungi including some non-Candida yeasts and non-Aspergillus moulds. In vivo data have shown IBX to be effective for treatment of candidiasis and aspergillosis. Ibrexafungerp is effective for the treatment of acute vulvovaginal candidiasis in completed phase 3 clinical trials.
- ibrexafungerp (IBX)
- SCY-078
- MK-3118
- fungal cell wall
- glucan synthase inhibitor
- triterpenoid antifungal
- fungerp
1. Introduction
Antifungals that inhibit the biosynthesis of β-(1,3)-D-glucan, an important cell wall component of most fungi, the potential to exhibit potent broad-spectrum of activity [1,2]. These drugs target an enzyme, β-(1,3)-D-glucan synthase that is unique to lower eukaryotes, limiting their toxicity in humans [1,3]. The echinocandins were the first glucan synthase inhibitors approved for use in 2001 [4] and have broad-spectrum activity against most common fungal pathogens (Candida spp., Aspergillus spp.), except for Cryptococcus neoformans [5]. Despite their good efficacy in the treatment of invasive Candida infections and low toxicity, their use is limited to parenteral administration only [2,3]. Echinocandins have very high molecular masses of about 1200 kDa [2,6], possibly resulting in their poor oral absorption [3,7,8]. Furthermore, distribution of the first-generation echinocandins to the central nervous system, intraocular fluids, and urine is poor, mainly due to their high protein-binding capabilities (>99%) and high molecular masses [3,7,8]. Active research into new drugs by high throughput screening of natural products from endophytic fungi led to the discovery of enfumafungin, a triterpene glycoside [9]. Enfumafungin is structurally distinct from echinocandins (Figure 1) [10,11], forming a new class of antifungals called “fungerps” (Antifungal Triterpenoid) [12–14]. Modifications of enfumafungin for improved oral bioavailability and pharmacokinetic properties led to the development of the semi-synthetic derivative, which was named ibrexafungerp (IBX) [15] by the World Health Organization’s international non-proprietary name group [16].
2. Mechanism of Action and Resistance
Ibrexafungerp (formerly SCY-078 or MK-3118) is a first-in-class triterpenoid antifungal that inhibits biosynthesis of β-(1,3)-D-glucan in the fungal cell wall. Glucan represents 50–60% of the fungal cell wall dry weight [17]. β-(1,3)-D-glucan is the most important component of the fungal wall, as many structures are covalently linked to it [17]; furthermore, it is the most abundant molecule in many fungi (65–90%) [17,18], making it an important antifungal target [1,12]. Inhibition of β-(1,3)-D-glucan biosynthesis compromises the fungal cell wall by making it highly permeable, disrupting osmotic pressure, which can lead to cell lysis [19–21]. β-(1,3)-D-glucan synthase is a transmembrane glycosyltransferase enzyme complex comprised of a catalytic Fks1p subunit encoded by the homologous genes FKS1 and FKS2 [22] and a third gene, FKS3 [23]; a rho GTPase regulatory subunit encoded by the Rho1p gene [24]. The catalytic unit binds UDP-glucose and the regulatory subunit binds GTP to catalyse the polymerization of UDP-glucose to β-(1,3)-D-glucan [25], which is incorporated into the fungal cell wall, where it functions mainly to maintain the structural integrity of the cell wall [19–21].
Ibrexafungerp (IBX) has a similar mechanism of action to the echinocandins [26,27] and acts by non-competitively inhibiting the β-(1,3) D-glucan synthase enzyme [12,27]. As with echinocandins, IBX has a fungicidal effect on Candida spp. [28] and a fungistatic effect on Aspergillus spp. [29,30]. However, the ibrexafungerp and echinocandin-binding sites on the enzyme are not the same, but partially overlap resulting in very limited cross-resistance between echinocandin- and ibrexafungerp-resistant strains [26,27,31]. Resistance to echinocandins is due to mutations in the FKS genes, encoding for the catalytic site of the β-(1,3) D-glucan synthase enzyme complex; specifically, mutations in two areas designated as hot spots 1 and 2 [32,33], have been associated with reduced susceptibility to echinocandins [33,34]. The β-(1,3) D-glucan synthase enzyme complex is critical for fungal cell wall activity; alterations of the catalytic core are associated with a decrease in the enzymatic reaction rate, causing slower β-(1,3) D-glucan biosynthesis [35]. Widespread use and prolonged courses of echinocandins have led to echinocandin resistance in Candida spp., especially C. glabrata and C. auris [36–40]. Ibrexafungerp has potent activity against echinocandin-resistant (ER) C. glabrata with FKS mutations [41], although certain FKS mutants have increased IBX MIC values, leading to 1.6–16-fold decreases in IBX susceptibility, compared to the wild-type strains [31]. Deletion mutations in the FKS1 (F625del) and FKS2 genes (F659del) lead to 40-fold and >121-fold increases in the MIC50 for IBX, respectively [31]. Furthermore, two additional mutations, W715L and A1390D, outside the hotspot 2 region in the FKS2 gene, resulted in 29-fold and 20-fold increases in the MIC50 for IBX, respectively [31]. The majority of resistance mutations to IBX in C. glabrata are located in the FKS2 gene [31,40], consistent with the hypothesis that biosynthesis of β-(1,3) D-glucan in C. glabrata is mostly mediated through the FKS2 gene [32].
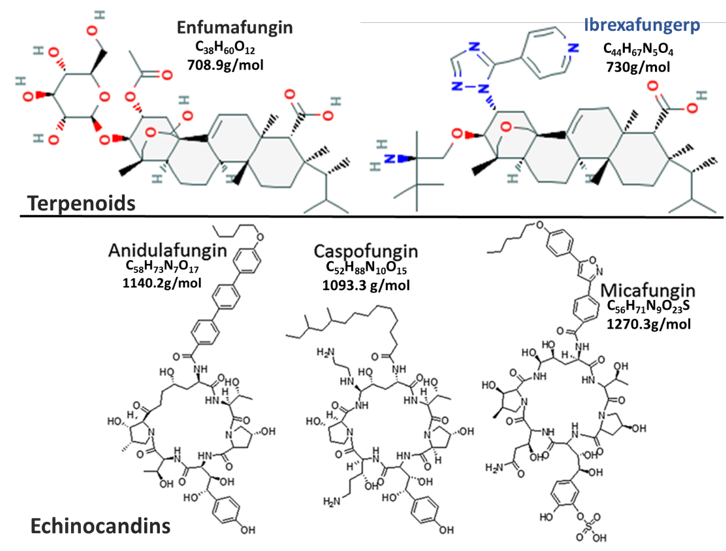
Figure 1. This is a figure comparing Fungerp and Echinocandin chemical structures (modified from [10,11]).
3. Important Pathogenic Fungi and Antifungal Spectrum
Invasive fungal infections (IFIs) are usually opportunistic [42]. The incidence of IFIs has been increasing globally due to a rise in immunocompromised populations, such as transplant recipients receiving immunosuppressive drugs; cancer patients on chemotherapy, people living with HIV/AIDS with low CD4 T-cell counts; patients undergoing major surgery and premature infants [42,43]. IFIs are a major cause of global mortality with approximately 1.5 million deaths per annum [44]; mainly due to Candida, Aspergillus, Pneumocystis, and Cryptococcus species [44]. Furthermore, there is an increase in antifungal resistance limiting available treatment options [45,46]; a shift in species causing invasive disease [47–50] to those that may be intrinsically resistant to some antifungals [51,52]. Several fungal pathogens (e.g., Candida auris, Histoplasma capsulatum, Cryptococcus spp., Emergomyces spp.) are gaining importance, especially in middle-income countries such as South Africa, India, Brazil and Colombia.
Candida auris has been reported in over 39 countries as an important emerging fungal pathogen [48] with a high crude mortality rate and a propensity for multidrug resistance [53–59]. C. auris has also been reported as an important cause of nosocomial outbreaks [60,61] due to its ability to colonize skin, form biofilms and resist standard disinfectants; due to its ease of person-to-person and person-to-environment transmission [60–62]. Within the last decade, C. auris became the third most common cause of candidaemia in South Africa, causing >10% of all culture-confirmed cases of invasive candidiasis [49,63,64]. A large proportion of C. auris infections are fatal due to the comorbidities in these patients, but multidrug- or even pan-resistance to available antifungals may also contribute to inappropriate treatment and adverse outcomes [53–59].
Invasive aspergillosis, a life-threatening acute disease, has a reported mortality of up to 85% [65,66]. First-line treatment is with voriconazole, though resistance to the azole class of drugs has been reported [67–69]. Resistance to amphotericin B formulations, used as alternative therapy, is rarer, although Aspergillus terreus is intrinsically amphotericin B-resistant [47].
Pneumocystis infections [70] have gained importance in the human immunodeficiency virus (HIV) era as an acquired immunodeficiency syndrome (AIDS)-related opportunistic infection [70]. Pneumocystis jirovecii causes Pneumocystis pneumonia in humans (PCP), which is still a leading cause of opportunistic infection in HIV/AIDS patients, even in the era of combination antiretroviral therapy [71]. In addition, Pneumocystis infections are increasing in non HIV-infected populations with impaired cell–mediated immunity, including those on immunomodulatory drugs or with underlying medical conditions such as inflammatory or autoimmune diseases [72,73]. First-line treatment is usually with trimethoprim-sulfamethoxazole (TMP-SMX) (alternatives include clindamycin-primaquine, atovaquone and pentamidine) [74] instead of the known antifungal classes. Pneumocystis jirovecii utilizes cholesterol, a mammalian-associated sterol, instead of ergosterol [75]; whose biosynthetic pathway is exploited by most antifungals, leading to intrinsic resistance of Pneumocystis spp. to these drugs [74]. Resistance mutations in the dihydropteroate synthase and cytochrome bc1 genes against TMP-SMX and atovaquone have already been identified [76]. Pneumocystis spp. are sensitive to glucan synthase inhibitors; however, owing to their unique life cycle, only the ascus (cyst) forms and not the trophic forms are sensitive to these drugs [74]. Glucan synthase inhibitors can therefore, only control, but not eradicate PCP colonization/infection [74].
The activity of glucan synthase inhibitors depends on the proportion of β-(1,3) D-glucan in the fungal cell wall, which can differ in different fungal species [77]. Most Saccharomyces, Candida and Aspergillus species, are susceptible to glucan synthase inhibitors [7,20,26,41,78–80], because β-(1,3) D-glucan is dominant in their cell walls [77]. These drugs also have activity against the ascus form of Pneumocystis jirovecii [81]. Fungi, such as those in the order Mucorales, Fusarium spp. and Scedosporium spp. with limited or no β-(1,3) D-glucan, are intrinsically resistant to this class of drugs [82]. However, a paradox occurs in Cryptococcus neoformans whose cell wall contains predominantly β-(1,3) D-glucan, yet is tolerant to this class of drugs [5,83].
4. In vitro activity
Yeasts: In vitro analysis of ibrexafungerp showed potent activity against a broad spectrum of >1300 Candida isolates [41,80,84-91]. Activity against C. lusitaniae and C. krusei (MIC range: 1-2 and 0.5-4 µg/mL, respectively), seemed to be less potent compared to other Candida spp. [87]. Compared to echinocandins, IBX was generally less potent (higher MIC values) for most common Candida spp., except for C. parapsilosis [86,87]. Ibrexafungerp showed strong activity against azole-resistant isolates (including C. albicans, C. parapsilosis, C. tropicalis, C. auris, C. krusei, C. glabrata, C. guilliermondii, C. lusitaniae, C. inconspicua) [84,87,90]; however, activity against echinocandin-resistant FKS mutants (C. albicans, C. krusei, C. tropicalis, C. glabrata, C. auris) was variable [84,86]. IBX has more activity against a majority of FKS mutants compared to the echinocandins, with 70-86% of echinocandin-resistant mutants susceptible to IBX compared to 17-50% for the echinocandins [80,84,86-89], possibly because selection of these mutants were due to echinocandin exposure. Most Candida echinocandin-resistant FKS mutants were susceptible to IBX [26,84,87,88], especially C. glabrata [41,87,89] and C. auris isolates [57,90], but some mutants with the F641S, F649del, F658del and F659del mutations had reduced susceptibility to IBX [80,84,86,89]. Ibrexafungerp produced enhanced activity against echinocandin-resistant C. albicans and C. glabrata, compared to caspofungin [80]. IBX also demonstrated potent activity against pan-resistant (resistance to ≥2 azoles, all echinocandins and amphotericin B) C. auris isolates from a New York City outbreak [57]. Candida biofilms use multiple resistance mechanism to escape from antifungals; leading to inherent resistance to azoles [92,93]. β-(1,3)-D-glucan is a key component of biofilm constituent; thus it is not surprising that IBX has shown activity against different biofilm-producing Candida species (C. albicans, C. parapsilosis, C. tropicalis, C. glabrata) [41,84].
Among 13 other non-Candida yeasts including Rhodotorula mucilaginosa, Trichosporon spp. (T. asahii, T. dermatis, T. inkin, T. japonicum) and Arxula adeninivorans, IBX activity was variable ranging from 0.25–≥128 µg/mL[84]. In another study of 100 non-Candida yeasts, IBX showed activity against Malassezia pachydermatis (MIC: 0.5 μg/mL), Pichia spp. (MIC: 0.5-1 μg/mL) and to some Trichosporon mucoides (MIC range: 0.125-2 μg/mL) [94]. In vitro analysis of IBX, at different pH levels against Candida spp., showed increased potency at lower pH, with MIC90 values of 0.5, 0.25 and <0.016 μg/mL at pH levels of 7.0, 5.72 and 4.5, respectively; indicating increased activity at low pH (owing to its pH-dependent solubility) that may increase potency for treatment of vulvovaginitis [95].
Moulds: Glucan synthase inhibitors have a fungistatic effect on some moulds [29,30], such as Aspergillus spp., despite high proportion of β-(1,3) D-glucan in the Aspergillus cell wall [29]. While these drugs may not kill these species of mould, they can have a profound antifungal effect both in vitro and in vivo [29,30]. Treatment with glucan synthase inhibitors causes lysis of growing hyphal tips; leading to short, stubby, highly branched hyphae or abnormal hyphal growth [29,96]. The fungistatic effect of these drugs means that MIC values are not accurate endpoint measures; instead minimum effective concentrations (MEC) (the lowest concentration at which abnormal hyphal growth occurs) are usually used in antifungal susceptibility testing [96]. IBX showed potent in vitro inhibition of Aspergillus species complexes (A. fumigatus, A. niger, A. flavus, A. terreus, A. nidulans, A. glaucus, A. ustus, A. versicolor, A. westerdijkiae, A. tamarii, A. calidoustus), including azole-resistant strains [26,30,80,94]. Echinocandin-resistant (ER) Aspergillus spp. are very rare, although an A. fumigatus mutant (S678P) has been described [97]. IBX showed increased activity against the S678P ER mutant with a MIC value that was 133-fold less than that observed for caspofungin [26].
IBX activity against medically important non-Aspergillus moulds including the Order Mucorales (Rhizopus, Mucor, Rhizomucor, Cunninghamella, Lichtheimia species), Fusarium spp., Scedosporium spp., Paecilomyces spp., and Scopulariopsis spp. showed variable results [98]. IBX was very active against Paecilomyces variotii, Penicillium citrinum, Scytalidium dimidiatum, Alternaria spp. and Cladosporium spp.; but had limited to no activity against the Mucorales, Fusarium spp., Purpureocillium lilacinum, Lichtheimia coerulea, Lichtheimia corymbifera, Acremonium spp., Cladosporium cladosporioides, Trichoderma citrinoviride and Trichoderma longibrachiatum [94,98]. IBX showed variable activity against Scopulariopsis spp. and modest activity against Scedosporium apiospermum and Lomentospora (formerly Scedosporium) prolificans [98]. Interestingly, IBX was the only drug, amongst those tested, that had any activity against the pan-resistant Lomentospora prolificans isolates [98].
Other fungi: Among dermatophytes, IBX demonstrated potent activity against Microsporum canis, Trichophyton tonsurans, Trichophyton mentagrophytes, and Trichophyton rubrum [94].
5. In vivo data from animal models
In a neutropenic murine model of invasive candidiasis, ibrexafungerp administered orally every 12 hours showed in vivo activity against both wild type (30 mg/kg) and echinocandin-resistant (40 mg/kg) C. glabrata strains, while caspofungin showed activity against the wild type strains only [99]. IBX given orally showed activity against C. albicans, C. glabrata, and C. parapsilosis in a neutropenic murine model of disseminated candidiasis, [100,101]. In an in vivo study of C. auris skin colonization in guinea pigs, 10 mg/kg oral dose of IBX produced a significantly lower fungal burden in the treated guinea pigs compared to those untreated; however with higher IBX doses (20 and 30 mg/kg) and with micafungin (5 mg/kg), the reduction in fungal burden was not significant [102]. At the end of treatment, histopathology results showed no fungal elements in all treated animals, while the untreated animals had fungal elements present, indicating that all treatment arms were able to clear the C. auris colonization [102]. Intra-abdominal candidiasis (IAC) is a difficult-to-treat invasive disease due to poor drug penetration at the site of infection and hence associated with high mortality [103,104]. In a mouse model of IAC, ibrexafungerp exhibited good penetration with robust accumulation within intra-abdominal lesions [103]. IBX concentrations in liver abscesses were 100-fold higher compared to that in serum [104].
IBX also demonstrated potent activity against both wild-type and azole-resistant strains of A. fumigatus in a murine model of invasive aspergillosis; with significant increase in survival and corresponding reductions in fungal kidney burden and serum galactomannan levels in treated mice compared to untreated mice [105]. Intravenous IBX (7.5 mg/kg/day) in combination with oral isavuconazole (40 mg/kg/day) showed potent activity in a neutropenic model of rabbit invasive pulmonary aspergillosis [106]. This drug combination demonstrated increased survival, reduced fungal pathogen-mediated pulmonary injury, decreased galactomannan antigenemia and serum (1,3)-β-D-glucan levels compared to either drug alone [106].
A prophylactic murine model of Pneumocystis pneumonia (PCP) found that oral IBX (30 mg/kg BID) reduced total nuclei and asci counts in lung tissue and improved survival; similar results were obtained with TMP/SMX, the gold standard for PCP therapy [81]. IBX reduced number of asci significantly by day 7 with asci being microscopically undetectable by day 14, in a therapeutic murine model of Pneumocystis pneumonia [107]. However, compared to TMP/SMX, total nuclei was only reduced, but was still detectable in the IBX group; and survival was better for the TMP/SMX group [107].
6. Clinical efficacy
Currently, there are 12 listed clinical trials for ibrexafungerp (Table 1), eight of which have been completed (https://ClinicalTrials.gov/; accessed 8 January 2021). Clinical data from at least 1000 participants using both single and multiple daily doses of IBX, as high as 1600 mg, revealed a safe and tolerable profile [108-111]. Mild adverse events were reported including headaches and gastrointestinal issues such as, diarrhea, nausea, vomiting and abdominal discomfort [108-111].
Table 1: This is a table showing the details of current clinical trials involving ibrexafungerp.
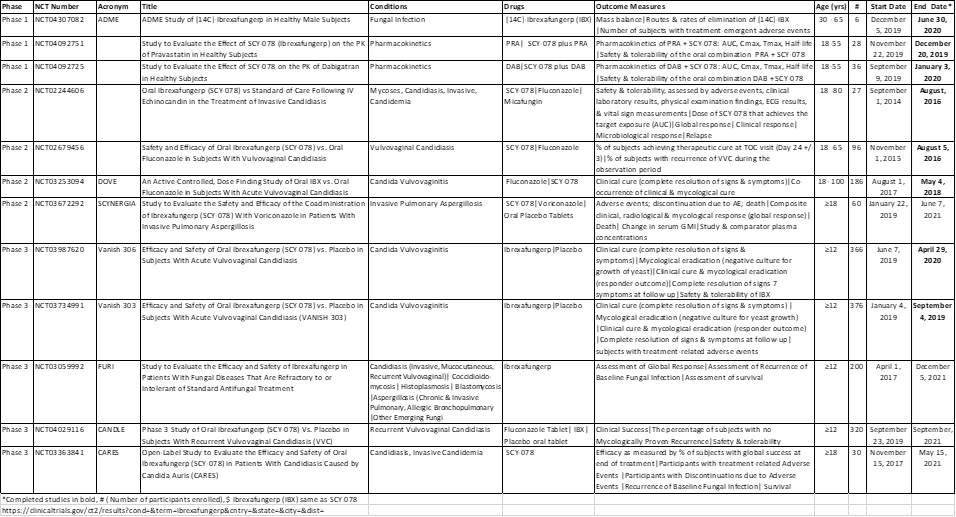
In a prospective Phase 2 (NCT02244606) randomized, open-label, multi-centre study in patients with invasive candidiasis including candidaemia, IBX administered as an oral step-down treatment after echinocandin therapy, was compared to the standard of care (SOC) treatments: either oral fluconazole or intravenous micafungin for fluconazole-resistant isolates [108]. Efficacy was determined by assessment of global response, with a favourable global response defined as resolution of signs and symptoms (clinical response) and negative Candida cultures (microbiological response), evaluated at the end of therapy [108]. The global response was similar between the IBX (500mg: 71%; 750mg: 86%) and SOC (75%) arms, although 750mg of IBX gave a higher response rate [108]. An ongoing, open-label, single-arm, phase 3 study (CARES: NCT03363841), expected to end in May 2021, is evaluating the efficacy of IBX in patients with Candida auris infections. In preliminary results, infection were completely resolved (culture negative) in 2 cases after treatment with IBX, including a case with difficult-to-treat C. auris that persisted after treatment with fluconazole and micafungun [112].
A phase 2, randomized, double-blind, double-dummy, dose-finding study (DOVE: NCT03253094) was done to compare the efficacy of oral ibrexafungerp to oral fluconazole (FLU) in patients with acute vulvovaginal candidiasis (VVC) [113]. The primary endpoints were clinical cure (complete resolution of all signs and symptoms) and mycological eradication (negative culture for yeast) at the test of cure (TOC) visit on day 10 [113]. The clinical cure (52% vs. 58%) and mycological eradication (63% vs. 63%) rates were similar for IBX and FLU, respectively; however, after 25 days, clinical cure (70% vs. 50%) and mycological eradication (48% vs 38%) rates were higher for IBX compared to FLU, respectively [113]. In two phase 3 randomized, double-blind, placebo-controlled clinical trials in patients with acute vulvovaginal candidiasis, VANISH 303 (NCT03734991) and VANISH 306 (NCT03987620), with the same end points as the DOVE study, complete resolution of all vaginal signs and symptoms by test of cure (day 10) date was significantly higher in the IBX groups compared to placebo [114,115]. In VANISH 303, clinical cure, mycological eradication, clinical improvement at TOC date and complete resolution of symptoms at day 25 were 51% vs. 29%, 50% vs. 19%, 64% vs. 37%, and 60% vs. 45%, respectively, in the IBX group compared to the placebo [114]. Similarly in VANISH 306, clinical cure, mycological eradication, clinical improvement and resolution of symptoms were 63% vs. 44%, 59% vs. 30%, 72% vs. 55%, and 74% vs. 52%, respectively, in the IBX group compared to the placebo [115]. A large (320 participants) multicentre, randomized, double-blind phase 3 study (CANDLE: NCT04029116) to investigate the efficacy of IBX compared placebo in participants with recurrent vulvovaginal candidiasis is currently ongoing and expected to end in September 2021 [116].
A phase 3 open-label single-arm study (FURI: NCT03059992) is investigating the efficacy of IBX in patients, with Candida and Aspergillus disease, who are either intolerant of or refractory to standard of care antifungal treatment [117]. The primary end-point is global success, defined as composite assessment of clinical, microbiological, serological and/or radiological responses at end of treatment [117]. Preliminary data have shown that the majority of the patients (83%) had either a complete or partial response (56%) or stable disease (27%), but 15% of the patients did not respond to IBX and 2% were indeterminate [117]. The FURI study was expanded to include other fungal diseases such as coccidioidomycosis, histoplasmosis, blastomycosis and other emerging fungi (table 1).
7. Pharmacokinetics/pharmacodynamics
Ibrexafungerp is the first orally-available glucan synthase inhibitor, with about 50% bioavailability in animal models [110]. In vitro studies using caco-2 cell monolayers and 5μM of IBX gave an average permeability of 8.9 ± ×10−6 cm/s in the apical-to-basolateral direction, indicating good oral absorption [110]. In vivo efficacy data from 3 murine models of disseminated candidiasis produced potent activity against C. albicans after 7 days of twice-daily oral treatment with IBX; with a mean therapeutic exposure of 14.3 μM·h across all models [110]. In other animal models, IBX was well absorbed into plasma after oral dosing with bioavailability values of >51, 45, and 35%; and half-lives (t1/2) of ~8.3, 9.1, and 15.2 h, for mice, rats, and dogs, respectively [110]. After intravenous administration, systemic clearance values of 0.68, 0.44, and 0.45 liter/h/kg; half-life values of ~5.5, 8.7, and 9.3 hours; and volume of distribution values of 5.3, 4.7, and 5.1 liters/kg, were observed for mice, rats, and dogs, respectively [110]. In vitro assessment of metabolic stability of IBX utilizing rodent, dog, and human liver microsomes gave clearance rates of ≤11, ≤48, and 34 µL/min/mg, respectively; corresponding to clearance rates of <40, ≤69, and 38 µL/min/kg, respectively, when scaled for in vivo intrinsic clearance [101]. Due to the observed long half-life and moderate liver clearance, a once-daily dosage was suggested for clinical trials [101]. For all species, declines in plasma concentrations were linear [110].
In vitro solubility of IBX is inversely correlated with pH; there was good solubility in simulated gastric fluid (SGF; >5.2 mg/ml) and fed-state intestinal fluid (FeSSIF; >3.0 mg/ml), but IBX was only slightly soluble in fasted-state simulated intestinal fluid (FaSSIF). The citrate formulation of IBX provided substantial improvement in solubility, increasing solubility in SGF and FeSSIF to >20 mg/mL and in FaSSIF to >4.2 mg/mL after 24hours [110]. IBX is a lipophilic compound; as such, observed protein binding was very high, similar to the echinocandins [101]. In vitro protein binding and blood-to-plasma ratio in mouse, rat, dog, and human cells gave very high protein binding, ranging from 99.8 to 99.5% [110]. Investigation of tissue distribution in the murine model of invasive candidiasis showed high volume distribution to the kidneys with area under the plasma concentration - time of zero to infinity (AUC0–∞) and maximum concentration (Cmax) values 20 to 25-fold greater than those for plasma [110]. Distribution volume is inversely proportional to affinity binding of proteins. The high distribution volumes observed for IBX indicate that while protein binding is high, it is most likely of low affinity; allowing better tissue distribution [110]. Furthermore, IBX is bound mainly to plasma proteins with a blood-to-plasma ratio ranging from 0.5 to 0.7; and not erythrocytes, giving IBX excellent properties for treating invasive disease [110]. Utilization of radioactive IBX ([14C]SCY-078), orally or parentally, in albino and pigmented rats, showed extensive distribution to tissues involved in invasive fungal disease, including kidney, lung, liver, spleen, bone marrow, muscle, vaginal tissue, and skin [118]. Tissue-to-blood AUC ratios after oral administration were several times higher in tissue relative to blood; with 54-fold higher concentrations in spleen; 50-fold higher in liver; 31-fold higher in lung; 25-fold higher in bone marrow; 20-fold higher in kidney; 12-fold higher in non-pigmented skin; 18-fold higher in pigmented skin; 9-fold higher in vaginal tissue; and 4-fold higher in skeletal muscle [118]. There was limited to no distribution to central nervous system tissues (brain and spinal cord); limited distribution to adipose tissues; and variable distribution to the eye (none to the lens, but very well distributed to the uvea) [118]. IBX elimination was shown to be mainly via feces and bile (∼90%); and a very small proportion via urine (∼1.5%) [118], probably due to high protein binding [110].
Investigation of cytochrome P450 (CYP) inhibition of IBX showed that it is a substrate for CYP3A4 and an inhibitor of CYP2C8; but has very little effect on other CYP isoforms (IC50 values >25μM for 1A2, 2B6, 2C9, 2C19, 2D6) [Wring SA, Park SM, unpublished data] [109,119]. A phase 1, open-label, 2-period crossover study, using a rosiglitazone, a sensitive substrate of CYP2C8 metabolism demonstrated that co-administration of IBX with rosiglitazone did not affect the maximum concentration values for rosiglitazone indicating that there is limited to no inhibition of CYP2C8 [109]. In another phase 1 study, the potential drug-drug interaction between IBX and tacrolimus, a substrate of CYP3A4 as well as a potent immunosuppressive drug used to prevent transplant rejection [120], was assessed [110]. The resultant PK values (AUC0-∞: 1.42-fold and Cmax: 1.03-fold) for IBX with tacrolimus or alone were similar, indicating that there was very little interaction between IBX and tacrolimus at therapeutic levels of IBX [110]. However, phase 1 studies using ketoconazole (strong CYP3A inhibitor) and diltiazem (moderate CYP3A4 inhibitor), found moderate to severe effects on IBX (AUC0-∞: 5.7-fold, Cmax: 2.5-fold) for ketoconazole and for moderate effects for diltiazem (AUC0-∞: 2.5-fold, Cmax: 2.2-fold) [119]. Taken together, these phase 1 studies indicate that IBX has limited potential for interaction with drugs metabolized by cytochrome P450; however, a dose adjustment may be necessary for potent CYP3A4 inhibitors [109,110,119].
8. Indications and usage
Most clinical trials have focused on the oral formulation of ibrexafungerp [87]. The use of ibrexafungerp for the treatment for vulvovaginal candidiasis (VVC) and prevention of recurrence of VVC was investigated in six studies that include efficacy (Table 1) [113-116]. These studies have demonstrated a favourable safety and tolerability profile, as well as high efficacy in the context of VVC [113-115], which led to acceptance of a new drug application (NDA), by the US Food and Drug Administration (FDA), for the treatment of VVC using ibrexafungerp [121]. Furthermore, Qualified Infectious Disease Product (QIDP) and Fast Track designations were granted by the FDA for the treatment of VVC and prevention of recurrent VVC with ibrexafungerp [121]. Results from completed clinical trials or preliminary data from ongoing trials have shown inbrexafungerp to be effective for treatment of invasive candidiasis including C. auris [108,112]; and for use as salvage therapy for refractory fungal infections [117,122]. Treatment of invasive pulmonary aspergillosis as combination therapy with azoles was found to be effective in in vitro [30] and in vivo animal models [106]; however clinical trials are ongoing [117]. Apart from candidiasis and aspergillosis, IBX is being assessed in the ongoing phase 3 FURI trial, for treatment of other fungal infections such as coccidioidomycosis, histoplasmosis, and blastomycosis [117].
9. Conclusions
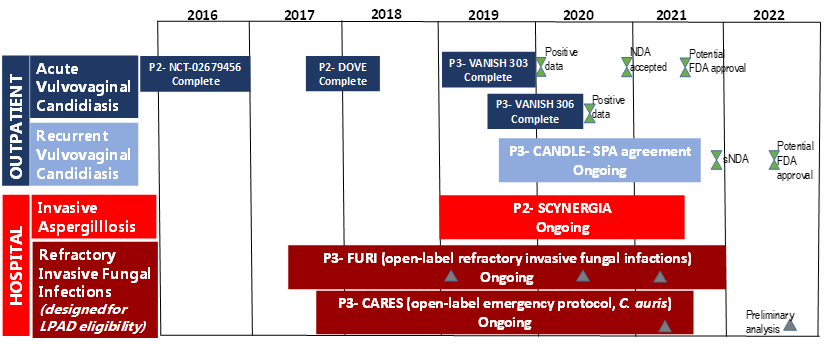
The rising incidence of IFIs, development of antifungal resistance and emergence of multi-drug resistant species, such as C. auris, are of public health concern. Thus, the development and availability of new drugs, such as ibrexafungerp that have activity against the most prevalent fungal pathogens, Candida and Aspergillus, including biofilm-forming strains, azole- and echinocandin-resistant strains provide alternative treatment options, where previously there were none or few options. The good oral bioavailability and once-daily dosing of ibrexafungerp will reduce the burden of IV administration, unnecessarily prolonged hospitalization, and complex dosing schedules, thereby increasing adherence and the likelihood of treatment success. Furthermore, IBX has favourable characteristics such as very low toxicities, enhanced activity at low pH for better activity in tissues and abscesses [104]; high tissue penetration for invasive disease; and low risk of drug-drug interactions that will allow treatment combinations and treatment of patients with multiple comorbidities [14]. IBX is indicated for vulvovaginal candidiasis, invasive candidiasis, invasive aspergillosis, pneumocystosis and some refractory invasive fungal infections [122]. The FDA has granted QIDP and fast track designations for oral and IV formulations of ibrexafungerp for the treatment of invasive candidiasis, vulvovaginal candidiasis and invasive aspergillosis; IBX was also given orphan drug designation for invasive candidiasis and invasive aspergillosis [122]. The FDA has set 1 June 2021 as the target date of action under the prescription drug user fee Act (figure 2) [14,122]; resulting in a commercial launch in the United States for treatment of VVC being planned in 2021 [123]. The high cost of new antifungals limits their access, especially in low- and middle-income countries (LMICs) where the fungal disease burden is high but the perceived commercial market is small, limiting manufacturers’ interest in additional regulatory approvals [124]. For example, echinocandins were registered in South Africa only 3-10 years after their approval and registration in the United States [4,125-127]. Ibrexafungerp patent applications are underway for 10 years of U.S. regulatory exclusivity plus composition-of-matter patent up to 2035, with additional applications pending, for a total of ~15 years of exclusivity in the U.S [123]. This will further delay access of this drug to most LMIC countries; hence early and efficient partnerships among pharmaceutical companies, governments and international organizations are necessary to promote global access of this novel medication.
Figure 2: This is a figure of the Ibrexafungerp clinical trials and milestones (modified from [14])
References
- Douglas, C.M. Fungal beta(1,3)-D-glucan synthesis. Med Mycol 2001, 39 Suppl 1, 55-66, doi:10.1080/mmy.39.1.55.66.
- Mikamo, H.; Sato, Y.; Tamaya, T. In vitro antifungal activity of FK463, a new water-soluble echinocandin-like lipopeptide. J Antimicrob Chemother 2000, 46, 485-487, doi:10.1093/jac/46.3.485.
- Denning, D.W. Echinocandin antifungal drugs. Lancet 2003, 362, 1142-1151, doi:10.1016/S0140-6736(03)14472-8.
- Cancidas (Caspofungin Acetate) Injection. Merck Research Laboratories. Application No.: 21-227. Approval Date: 1/26/2001. Food and Drug Administration, Center for Drug Evaluation and Research, Division of Special Pathogen and Immunologic Drug Products. [Online]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21227_Cancidas.cfm (29 December 2020, date last accessed).
- Feldmesser, M.; Kress, Y.; Mednick, A.; Casadevall, A. The effect of the echinocandin analogue caspofungin on cell wall glucan synthesis by Cryptococcus neoformans. J Infect Dis 2000, 182, 1791-1795, doi:10.1086/317614.
- Kurtz, M.B.; Rex, J.H. Glucan synthase inhibitors as antifungal agents. Adv Protein Chem 2001, 56, 423-475, doi:10.1016/s0065-3233(01)56011-8.
- Chandrasekar, P.H.; Sobel, J.D. Micafungin: a new echinocandin. Clin Infect Dis 2006, 42, 1171-1178, doi:10.1086/501020.
- Vazquez, J.A.; Sobel, J.D. Anidulafungin: a novel echinocandin. Clin Infect Dis 2006, 43, 215-222, doi:10.1086/505204.
- Pelaez, F.; Cabello, A.; Platas, G.; Diez, M.T.; Gonzalez del Val, A.; Basilio, A.; Martan, I.; Vicente, F.; Bills, G.E.; Giacobbe, R.A., et al. The discovery of enfumafungin, a novel antifungal compound produced by an endophytic Hormonema species biological activity and taxonomy of the producing organisms. Syst Appl Microbiol 2000, 23, 333-343, doi:10.1016/s0723-2020(00)80062-4.
- PubChem [Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; 2004-. PubChem Compound Summary [cited 2020 Dec. 30]. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/.
- Coad, B.R.; Lamont-Friedrich, S.J.; Gwynne, L.; Jasieniak, M.; Griesser, S.S.; Traven, A.; Peleg, A.Y.; Griesser, H.J. Surface coatings with covalently attached caspofungin are effective in eliminating fungal pathogens. J Mater Chem B 2015, 3, 8469-8476, doi:10.1039/c5tb00961h.
- Onishi, J.; Meinz, M.; Thompson, J.; Curotto, J.; Dreikorn, S.; Rosenbach, M.; Douglas, C.; Abruzzo, G.; Flattery, A.; Kong, L., et al. Discovery of novel antifungal (1,3)-beta-D-glucan synthase inhibitors. Antimicrob Agents Chemother 2000, 44, 368-377, doi:10.1128/aac.44.2.368-377.2000.
- Davis, M.R.; Donnelley, M.A.; Thompson, G.R. Ibrexafungerp: A novel oral glucan synthase inhibitor. Med Mycol 2020, 58, 579-592, doi:10.1093/mmy/myz083.
- Azie, N. A Phase 2b, Dose Finding Study Evaluating Oral Ibrexafungerp in Moderate to Severe Acute Vulvovaginal Candidiasis (DOVE). Oral presentation, 3rd ISIDOG Congress, Porto Portugal online @ https://www.scynexis.com/science/publications-and-presentations/posters-and-presentations?page=2 2019.
- Apgar, J.M.; Wilkening, R.R.; Parker, D.L., Jr.; Meng, D.; Wildonger, K.J.; Sperbeck, D.; Greenlee, M.L.; Balkovec, J.M.; Flattery, A.M.; Abruzzo, G.K., et al. Ibrexafungerp: An orally active beta-1,3-glucan synthesis inhibitor. Bioorg Med Chem Lett 2020, 10.1016/j.bmcl.2020.127661, 127661, doi:10.1016/j.bmcl.2020.127661.
- Scynexis. The World Health Organization Recognizes New Antifungal Class by Granting "ibrexafungerp" to SCYNEXIS as the International Non-Proprietary Name for SCY-078. SCYNEXIS, Inc. press release online @ https://ir.scynexis.com/press-releases/detail/156/the-world-health-organization-recognizes-new-antifungal 2018.
- Garcia-Rubio, R.; de Oliveira, H.C.; Rivera, J.; Trevijano-Contador, N. The Fungal Cell Wall: Candida, Cryptococcus, and Aspergillus Species. Front Microbiol 2019, 10, 2993, doi:10.3389/fmicb.2019.02993.
- Ruiz-Herrera, J.; Ortiz-Castellanos, L. Cell wall glucans of fungi. A review. Cell Surf 2019, 5, 100022, doi:10.1016/j.tcsw.2019.100022.
- Fleet, G.H. Composition and structure of yeast cell walls. Curr Top Med Mycol 1985, 1, 24-56, doi:10.1007/978-1-4613-9547-8_2.
- Deresinski, S.C.; Stevens, D.A. Caspofungin. Clin Infect Dis 2003, 36, 1445-1457, doi:10.1086/375080.
- Latge, J.P. The cell wall: a carbohydrate armour for the fungal cell. Mol Microbiol 2007, 66, 279-290, doi:10.1111/j.1365-2958.2007.05872.x.
- Mazur, P.; Morin, N.; Baginsky, W.; el-Sherbeini, M.; Clemas, J.A.; Nielsen, J.B.; Foor, F. Differential expression and function of two homologous subunits of yeast 1,3-beta-D-glucan synthase. Mol Cell Biol 1995, 15, 5671-5681, doi:10.1128/mcb.15.10.5671.
- Dijkgraaf, G.J.; Abe, M.; Ohya, Y.; Bussey, H. Mutations in Fks1p affect the cell wall content of beta-1,3- and beta-1,6-glucan in Saccharomyces cerevisiae. Yeast 2002, 19, 671-690, doi:10.1002/yea.866.
- Kondoh, O.; Tachibana, Y.; Ohya, Y.; Arisawa, M.; Watanabe, T. Cloning of the RHO1 gene from Candida albicans and its regulation of beta-1,3-glucan synthesis. J Bacteriol 1997, 179, 7734-7741, doi:10.1128/jb.179.24.7734-7741.1997.
- Shematek, E.M.; Braatz, J.A.; Cabib, E. Biosynthesis of the yeast cell wall. I. Preparation and properties of beta-(1 leads to 3)glucan synthetase. J Biol Chem 1980, 255, 888-894.
- Pfaller, M.A.; Messer, S.A.; Motyl, M.R.; Jones, R.N.; Castanheira, M. In vitro activity of a new oral glucan synthase inhibitor (MK-3118) tested against Aspergillus spp. by CLSI and EUCAST broth microdilution methods. Antimicrob Agents Chemother 2013, 57, 1065-1068, doi:10.1128/AAC.01588-12.
- Walker, S.S.; Xu, Y.; Triantafyllou, I.; Waldman, M.F.; Mendrick, C.; Brown, N.; Mann, P.; Chau, A.; Patel, R.; Bauman, N., et al. Discovery of a novel class of orally active antifungal beta-1,3-D-glucan synthase inhibitors. Antimicrob Agents Chemother 2011, 55, 5099-5106, doi:10.1128/AAC.00432-11.
- Scorneaux, B.; Angulo, D.; Borroto-Esoda, K.; Ghannoum, M.; Peel, M.; Wring, S. SCY-078 Is Fungicidal against Candida Species in Time-Kill Studies. Antimicrob Agents Chemother 2017, 61, doi:10.1128/AAC.01961-16.
- Bowman, J.C.; Hicks, P.S.; Kurtz, M.B.; Rosen, H.; Schmatz, D.M.; Liberator, P.A.; Douglas, C.M. The antifungal echinocandin caspofungin acetate kills growing cells of Aspergillus fumigatus in vitro. Antimicrob Agents Chemother 2002, 46, 3001-3012, doi:10.1128/aac.46.9.3001-3012.2002.
- Ghannoum, M.; Long, L.; Larkin, E.L.; Isham, N.; Sherif, R.; Borroto-Esoda, K.; Barat, S.; Angulo, D. Evaluation of the Antifungal Activity of the Novel Oral Glucan Synthase Inhibitor SCY-078, Singly and in Combination, for the Treatment of Invasive Aspergillosis. Antimicrob Agents Chemother 2018, 62, doi:10.1128/AAC.00244-18.
- Jimenez-Ortigosa, C.; Perez, W.B.; Angulo, D.; Borroto-Esoda, K.; Perlin, D.S. De Novo Acquisition of Resistance to SCY-078 in Candida glabrata Involves FKS Mutations That both Overlap and Are Distinct from Those Conferring Echinocandin Resistance. Antimicrob Agents Chemother 2017, 61, doi:10.1128/AAC.00833-17.
- Perlin, D.S. Resistance to echinocandin-class antifungal drugs. Drug Resist Updat 2007, 10, 121-130, doi:10.1016/j.drup.2007.04.002.
- Garcia-Effron, G.; Lee, S.; Park, S.; Cleary, J.D.; Perlin, D.S. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-D-glucan synthase: implication for the existing susceptibility breakpoint. Antimicrob Agents Chemother 2009, 53, 3690-3699, doi:10.1128/AAC.00443-09.
- Park, S.; Kelly, R.; Kahn, J.N.; Robles, J.; Hsu, M.J.; Register, E.; Li, W.; Vyas, V.; Fan, H.; Abruzzo, G., et al. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 2005, 49, 3264-3273, doi:10.1128/AAC.49.8.3264-3273.2005.
- Ben-Ami, R.; Garcia-Effron, G.; Lewis, R.E.; Gamarra, S.; Leventakos, K.; Perlin, D.S.; Kontoyiannis, D.P. Fitness and virulence costs of Candida albicans FKS1 hot spot mutations associated with echinocandin resistance. J Infect Dis 2011, 204, 626-635, doi:10.1093/infdis/jir351.
- Alexander, B.D.; Johnson, M.D.; Pfeiffer, C.D.; Jimenez-Ortigosa, C.; Catania, J.; Booker, R.; Castanheira, M.; Messer, S.A.; Perlin, D.S.; Pfaller, M.A. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis 2013, 56, 1724-1732, doi:10.1093/cid/cit136.
- Pham, C.D.; Iqbal, N.; Bolden, C.B.; Kuykendall, R.J.; Harrison, L.H.; Farley, M.M.; Schaffner, W.; Beldavs, Z.G.; Chiller, T.M.; Park, B.J., et al. Role of FKS Mutations in Candida glabrata: MIC values, echinocandin resistance, and multidrug resistance. Antimicrob Agents Chemother 2014, 58, 4690-4696, doi:10.1128/AAC.03255-14.
- Chowdhary, A.; Prakash, A.; Sharma, C.; Kordalewska, M.; Kumar, A.; Sarma, S.; Tarai, B.; Singh, A.; Upadhyaya, G.; Upadhyay, S., et al. A multicentre study of antifungal susceptibility patterns among 350 Candida auris isolates (2009-17) in India: role of the ERG11 and FKS1 genes in azole and echinocandin resistance. J Antimicrob Chemother 2018, 73, 891-899, doi:10.1093/jac/dkx480.
- Kordalewska, M.; Lee, A.; Park, S.; Berrio, I.; Chowdhary, A.; Zhao, Y.; Perlin, D.S. Understanding Echinocandin Resistance in the Emerging Pathogen Candida auris. Antimicrob Agents Chemother 2018, 62, doi:10.1128/AAC.00238-18.
- Naicker, S.D.; Magobo, R.E.; Zulu, T.G.; Maphanga, T.G.; Luthuli, N.; Lowman, W.; Govender, N.P. Two echinocandin-resistant Candida glabrata FKS mutants from South Africa. Med Mycol Case Rep 2016, 11, 24-26, doi:10.1016/j.mmcr.2016.03.004.
- Nunnally, N.S.; Etienne, K.A.; Angulo, D.; Lockhart, S.R.; Berkow, E.L. In Vitro Activity of Ibrexafungerp, a Novel Glucan Synthase Inhibitor against Candida glabrata Isolates with FKS Mutations. Antimicrob Agents Chemother 2019, 63, doi:10.1128/AAC.01692-19.
- Perfect, J.R. The antifungal pipeline: a reality check. Nat Rev Drug Discov 2017, 16, 603-616, doi:10.1038/nrd.2017.46.
- Pfaller, M.A.; Pappas, P.G.; Wingard, J.R. Invasive Fungal Pathogens: Current Epidemiological Trends. Clinical Infectious Diseases 2006, 43, S3-S14.
- Brown, G.D.; Denning, D.W.; Gow, N.A.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: human fungal infections. Sci Transl Med 2012, 4, 165rv113, doi:10.1126/scitranslmed.3004404.
- WHO. Antimicrobial resistance: global report on surveillance. Geneva, Switzerland: WHO, 2014. World Health Organization online @ https://www.who.int/drugresistance/documents/surveillancereport/en/ 2014.
- Arendrup, M.C. Update on antifungal resistance in Aspergillus and Candida. Clin Microbiol Infect 2014, 20 Suppl 6, 42-48, doi:10.1111/1469-0691.12513.
- Pfaller, M.A.; Diekema, D.J. Rare and emerging opportunistic fungal pathogens: concern for resistance beyond Candida albicans and Aspergillus fumigatus. J Clin Microbiol 2004, 42, 4419-4431, doi:10.1128/JCM.42.10.4419-4431.2004.
- Wickes, B.L. Analysis of a Candida auris Outbreak Provides New Insights into an Emerging Pathogen. J Clin Microbiol 2020, 58, doi:10.1128/JCM.02083-19.
- van Schalkwyk, E.; Mpembe, R.S.; Thomas, J.; Shuping, L.; Ismail, H.; Lowman, W.; Karstaedt, A.S.; Chibabhai, V.; Wadula, J.; Avenant, T., et al. Epidemiologic Shift in Candidemia Driven by Candida auris, South Africa, 2016-2017(1). Emerg Infect Dis 2019, 25, 1698-1707, doi:10.3201/eid2509.190040.
- Govender, N.P.; Patel, J.; Magobo, R.E.; Naicker, S.; Wadula, J.; Whitelaw, A.; Coovadia, Y.; Kularatne, R.; Govind, C.; Lockhart, S.R., et al. Emergence of azole-resistant Candida parapsilosis causing bloodstream infection: results from laboratory-based sentinel surveillance in South Africa. J Antimicrob Chemother 2016, 71, 1994-2004, doi:10.1093/jac/dkw091.
- Fridkin, S.K.; Jarvis, W.R. Epidemiology of nosocomial fungal infections. Clinical Microbiology Reviews 1996, 9, 499-511, doi:10.1128/CMR.9.4.499.
- Pfaller, M.A.; Messer, S.A.; Moet, G.J.; Jones, R.N.; Castanheira, M. Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in Intensive Care Unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program (2008-2009). Int J Antimicrob Agents 2011, 38, 65-69, doi:10.1016/j.ijantimicag.2011.02.016.
- Chowdhary, A.; Sharma, C.; Duggal, S.; Agarwal, K.; Prakash, A.; Singh, P.K.; Jain, S.; Kathuria, S.; Randhawa, H.S.; Hagen, F., et al. New clonal strain of Candida auris, Delhi, India. Emerg Infect Dis 2013, 19, 1670-1673, doi:10.3201/eid1910.130393.
- Chowdhary, A.; Anil Kumar, V.; Sharma, C.; Prakash, A.; Agarwal, K.; Babu, R.; Dinesh, K.R.; Karim, S.; Singh, S.K.; Hagen, F., et al. Multidrug-resistant endemic clonal strain of Candida auris in India. Eur J Clin Microbiol Infect Dis 2014, 33, 919-926, doi:10.1007/s10096-013-2027-1.
- Lockhart, S.R.; Etienne, K.A.; Vallabhaneni, S.; Farooqi, J.; Chowdhary, A.; Govender, N.P.; Colombo, A.L.; Calvo, B.; Cuomo, C.A.; Desjardins, C.A., et al. Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin Infect Dis 2017, 64, 134-140, doi:10.1093/cid/ciw691.
- Lockhart, S.R.; Jackson, B.R.; Vallabhaneni, S.; Ostrosky-Zeichner, L.; Pappas, P.G.; Chiller, T. Thinking beyond the Common Candida Species: Need for Species-Level Identification of Candida Due to the Emergence of Multidrug-Resistant Candida auris. J Clin Microbiol 2017, 55, 3324-3327, doi:10.1128/JCM.01355-17.
- Zhu, Y.C.; Barat, S.A.; Borroto-Esoda, K.; Angulo, D.; Chaturvedi, S.; Chaturvedi, V. Pan-resistant Candida auris isolates from the outbreak in New York are susceptible to ibrexafungerp (a glucan synthase inhibitor). Int J Antimicrob Agents 2020, 55, 105922, doi:10.1016/j.ijantimicag.2020.105922.
- Ostrowsky, B.; Greenko, J.; Adams, E.; Quinn, M.; O'Brien, B.; Chaturvedi, V.; Berkow, E.; Vallabhaneni, S.; Forsberg, K.; Chaturvedi, S., et al. Candida auris Isolates Resistant to Three Classes of Antifungal Medications - New York, 2019. MMWR Morb Mortal Wkly Rep 2020, 69, 6-9, doi:10.15585/mmwr.mm6901a2.
- Osei Sekyere, J. Candida auris: A systematic review and meta-analysis of current updates on an emerging multidrug-resistant pathogen. Microbiologyopen 2018, 7, e00578, doi:10.1002/mbo3.578.
- Sabino, R.; Verissimo, C.; Pereira, A.A.; Antunes, F. Candida auris, an Agent of Hospital-Associated Outbreaks: Which Challenging Issues Do We Need to Have in Mind? Microorganisms 2020, 8, doi:10.3390/microorganisms8020181.
- Chaabane, F.; Graf, A.; Jequier, L.; Coste, A.T. Review on Antifungal Resistance Mechanisms in the Emerging Pathogen Candida auris. Front Microbiol 2019, 10, 2788, doi:10.3389/fmicb.2019.02788.
- Forsberg, K.; Woodworth, K.; Walters, M.; Berkow, E.L.; Jackson, B.; Chiller, T.; Vallabhaneni, S. Candida auris: The recent emergence of a multidrug-resistant fungal pathogen. Med Mycol 2019, 57, 1-12, doi:10.1093/mmy/myy054.
- Govender, N.P.; Magobo, R.E.; Mpembe, R.; Mhlanga, M.; Matlapeng, P.; Corcoran, C.; Govind, C.; Lowman, W.; Senekal, M.; Thomas, J. Candida auris in South Africa, 2012-2016. Emerg Infect Dis 2018, 24, 2036-2040, doi:10.3201/eid2411.180368.
- Magobo, R.E.; Corcoran, C.; Seetharam, S.; Govender, N.P. Candida auris-associated candidemia, South Africa. Emerg Infect Dis 2014, 20, 1250-1251, doi:10.3201/eid2007.131765.
- Patterson, T.F.; Kirkpatrick, W.R.; White, M.; Hiemenz, J.W.; Wingard, J.R.; Dupont, B.; Rinaldi, M.G.; Stevens, D.A.; Graybill, J.R. Invasive aspergillosis. Disease spectrum, treatment practices, and outcomes. I3 Aspergillus Study Group. Medicine (Baltimore) 2000, 79, 250-260, doi:10.1097/00005792-200007000-00006.
- Lin, S.J.; Schranz, J.; Teutsch, S.M. Aspergillosis case-fatality rate: systematic review of the literature. Clin Infect Dis 2001, 32, 358-366, doi:10.1086/318483.
- Vermeulen, E.; Maertens, J.; De Bel, A.; Nulens, E.; Boelens, J.; Surmont, I.; Mertens, A.; Boel, A.; Lagrou, K. Nationwide Surveillance of Azole Resistance in Aspergillus Diseases. Antimicrob Agents Chemother 2015, 59, 4569-4576, doi:10.1128/AAC.00233-15.
- Snelders, E.; van der Lee, H.A.; Kuijpers, J.; Rijs, A.J.; Varga, J.; Samson, R.A.; Mellado, E.; Donders, A.R.; Melchers, W.J.; Verweij, P.E. Emergence of azole resistance in Aspergillus fumigatus and spread of a single resistance mechanism. PLoS Med 2008, 5, e219, doi:10.1371/journal.pmed.0050219.
- Snelders, E.; Huis In 't Veld, R.A.; Rijs, A.J.; Kema, G.H.; Melchers, W.J.; Verweij, P.E. Possible environmental origin of resistance of Aspergillus fumigatus to medical triazoles. Appl Environ Microbiol 2009, 75, 4053-4057, doi:10.1128/AEM.00231-09.
- Mitchell, T.G.; Perfect, J.R. Cryptococcosis in the era of AIDS--100 years after the discovery of Cryptococcus neoformans. Clin Microbiol Rev 1995, 8, 515-548, doi:10.1128/CMR.8.4.515-548.1995.
- Walzer, P.D.; Evans, H.E.; Copas, A.J.; Edwards, S.G.; Grant, A.D.; Miller, R.F. Early predictors of mortality from Pneumocystis jirovecii pneumonia in HIV-infected patients: 1985-2006. Clin Infect Dis 2008, 46, 625-633, doi:10.1086/526778.
- Duncan, R.A.; von Reyn, C.F.; Alliegro, G.M.; Toossi, Z.; Sugar, A.M.; Levitz, S.M. Idiopathic CD4+ T-lymphocytopenia--four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med 1993, 328, 393-398, doi:10.1056/NEJM199302113280604.
- Reid, A.B.; Chen, S.C.; Worth, L.J. Pneumocystis jirovecii pneumonia in non-HIV-infected patients: new risks and diagnostic tools. Curr Opin Infect Dis 2011, 24, 534-544, doi:10.1097/QCO.0b013e32834cac17.
- Porollo, A.; Meller, J.; Joshi, Y.; Jaiswal, V.; Smulian, A.G.; Cushion, M.T. Analysis of current antifungal agents and their targets within the Pneumocystis carinii genome. Curr Drug Targets 2012, 13, 1575-1585, doi:10.2174/138945012803530107.
- Kaneshiro, E.S.; Ellis, J.E.; Jayasimhulu, K.; Beach, D.H. Evidence for the presence of "metabolic sterols" in Pneumocystis: identification and initial characterization of Pneumocystis carinii sterols. J Eukaryot Microbiol 1994, 41, 78-85, doi:10.1111/j.1550-7408.1994.tb05938.x.
- Malamba, S.; Sandison, T.; Lule, J.; Reingold, A.; Walker, J.; Dorsey, G.; Mermin, J. Plasmodium falciparum dihydrofolate reductase and dihyropteroate synthase mutations and the use of trimethoprim-sulfamethoxazole prophylaxis among persons infected with human immunodeficiency virus. Am J Trop Med Hyg 2010, 82, 766-771, doi:10.4269/ajtmh.2010.08-0408.
- Chen, S.C.; Slavin, M.A.; Sorrell, T.C. Echinocandin antifungal drugs in fungal infections: a comparison. Drugs 2011, 71, 11-41, doi:10.2165/11585270-000000000-00000.
- Eschenauer, G.; Depestel, D.D.; Carver, P.L. Comparison of echinocandin antifungals. Ther Clin Risk Manag 2007, 3, 71-97, doi:10.2147/tcrm.2007.3.1.71.
- de la Torre, P.; Meyer, D.K.; Reboli, A.C. Anidulafungin: a novel echinocandin for candida infections. Future Microbiol 2008, 3, 593-601, doi:10.2217/17460913.3.6.593.
- Jimenez-Ortigosa, C.; Paderu, P.; Motyl, M.R.; Perlin, D.S. Enfumafungin derivative MK-3118 shows increased in vitro potency against clinical echinocandin-resistant Candida Species and Aspergillus species isolates. Antimicrob Agents Chemother 2014, 58, 1248-1251, doi:10.1128/AAC.02145-13.
- Cushion, M.; Ashbaugh, A.; Borroto-Esoda, K.; Barat, S.A.; Angulo, D. SCY-078 demonstrates antifungal activity against pneumocystis in a prophylactic murine model of pneumocystis pneumonia. ASM Microbe Online 2018.
- Espinel-Ingroff, A. Comparison of In vitro activities of the new triazole SCH56592 and the echinocandins MK-0991 (L-743,872) and LY303366 against opportunistic filamentous and dimorphic fungi and yeasts. J Clin Microbiol 1998, 36, 2950-2956, doi:10.1128/JCM.36.10.2950-2956.1998.
- Maligie, M.A.; Selitrennikoff, C.P. Cryptococcus neoformans resistance to echinocandins: (1,3)beta-glucan synthase activity is sensitive to echinocandins. Antimicrob Agents Chemother 2005, 49, 2851-2856, doi:10.1128/AAC.49.7.2851-2856.2005.
- Marcos-Zambrano, L.J.; Gomez-Perosanz, M.; Escribano, P.; Bouza, E.; Guinea, J. The novel oral glucan synthase inhibitor SCY-078 shows in vitro activity against sessile and planktonic Candida spp. J Antimicrob Chemother 2017, 72, 1969-1976, doi:10.1093/jac/dkx010.
- Larkin, E.; Hager, C.; Chandra, J.; Mukherjee, P.K.; Retuerto, M.; Salem, I.; Long, L.; Isham, N.; Kovanda, L.; Borroto-Esoda, K., et al. The Emerging Pathogen Candida auris: Growth Phenotype, Virulence Factors, Activity of Antifungals, and Effect of SCY-078, a Novel Glucan Synthesis Inhibitor, on Growth Morphology and Biofilm Formation. Antimicrob Agents Chemother 2017, 61, doi:10.1128/AAC.02396-16.
- Pfaller, M.A.; Messer, S.A.; Rhomberg, P.R.; Borroto-Esoda, K.; Castanheira, M. Differential Activity of the Oral Glucan Synthase Inhibitor SCY-078 against Wild-Type and Echinocandin-Resistant Strains of Candida Species. Antimicrob Agents Chemother 2017, 61, doi:10.1128/AAC.00161-17.
- Schell, W.A.; Jones, A.M.; Borroto-Esoda, K.; Alexander, B.D. Antifungal Activity of SCY-078 and Standard Antifungal Agents against 178 Clinical Isolates of Resistant and Susceptible Candida Species. Antimicrob Agents Chemother 2017, 61, doi:10.1128/AAC.01102-17.
- Berkow, E.L.; Angulo, D.; Lockhart, S.R. In Vitro Activity of a Novel Glucan Synthase Inhibitor, SCY-078, against Clinical Isolates of Candida auris. Antimicrob Agents Chemother 2017, 61, doi:10.1128/AAC.00435-17.
- Ghannoum, M.; Long, L.; Isham, N.; Hager, C.; Wilson, R.; Borroto-Esoda, K.; Barat, S.; Angulo, D. Activity of a novel 1,3-beta-D-glucan Synthase Inhibitor, Ibrexafungerp (formerly SCY-078), Against Candida glabrata. Antimicrob Agents Chemother 2019, 10.1128/AAC.01510-19, doi:10.1128/AAC.01510-19.
- Arendrup, M.C.; Jorgensen, K.M.; Hare, R.K.; Chowdhary, A. In Vitro Activity of Ibrexafungerp (SCY-078) against Candida auris Isolates as Determined by EUCAST Methodology and Comparison with Activity against C. albicans and C. glabrata and with the Activities of Six Comparator Agents. Antimicrob Agents Chemother 2020, 64, doi:10.1128/AAC.02136-19.
- Zhu, Y.C.; Barat, S.A.; Borroto-Esoda, K.; Angulo, D.; Chaturvedi, S.; Chaturvedi, V. In vitro Efficacy of Novel Glucan Synthase Inhibitor, Ibrexafungerp (SCY-078), Against Multidrug- and Pan-resistant Candida auris Isolates from the Outbreak in New York. BioRxiv 2020, doi: https://doi.org/10.1101/811182 doi:doi: https://doi.org/10.1101/811182
- Chandra, J.; Mukherjee, P.K.; Leidich, S.D.; Faddoul, F.F.; Hoyer, L.L.; Douglas, L.J.; Ghannoum, M.A. Antifungal resistance of candidal biofilms formed on denture acrylic in vitro. J Dent Res 2001, 80, 903-908, doi:10.1177/00220345010800031101.
- Cowen, L.E.; Sanglard, D.; Howard, S.J.; Rogers, P.D.; Perlin, D.S. Mechanisms of Antifungal Drug Resistance. Cold Spring Harb Perspect Med 2014, 5, a019752, doi:10.1101/cshperspect.a019752.
- Ghannoum, M.; Long, L.; Sherif, R.; Abidi, F.Z.; Borroto-Esoda, K.; Barat, S.; Angulo, D.; Wiederhold, N. Determination of Antifungal Activity of SCY-078, a Novel Glucan Synthase Inhibitor, Against a broad panel of Rare Pathogenic Fungi ASM Microbe Online 2020.
- Larkin, E.L.; Long, L.; Isham, N.; Borroto-Esoda, K.; Barat, S.; Angulo, D.; Wring, S.; Ghannoum, M. A Novel 1,3-Beta-d-Glucan Inhibitor, Ibrexafungerp (Formerly SCY-078), Shows Potent Activity in the Lower pH Environment of Vulvovaginitis. Antimicrob Agents Chemother 2019, 63, doi:10.1128/AAC.02611-18.
- Odds, F.C.; Brown, A.J.; Gow, N.A. Antifungal agents: mechanisms of action. Trends Microbiol 2003, 11, 272-279, doi:10.1016/s0966-842x(03)00117-3.
- Rocha, E.M.; Garcia-Effron, G.; Park, S.; Perlin, D.S. A Ser678Pro substitution in Fks1p confers resistance to echinocandin drugs in Aspergillus fumigatus. Antimicrob Agents Chemother 2007, 51, 4174-4176, doi:10.1128/AAC.00917-07.
- Lamoth, F.; Alexander, B.D. Antifungal activities of SCY-078 (MK-3118) and standard antifungal agents against clinical non-Aspergillus mold isolates. Antimicrob Agents Chemother 2015, 59, 4308-4311, doi:10.1128/AAC.00234-15.
- Wiederhold, N.P.; Najvar, L.K.; Jaramillo, R.; Olivo, M.; Pizzini, J.; Catano, G.; Patterson, T.F. Oral glucan synthase inhibitor SCY-078 is effective in an experimental murine model of invasive candidiasis caused by WT and echinocandin-resistant Candida glabrata. J Antimicrob Chemother 2018, 73, 448-451, doi:10.1093/jac/dkx422.
- Lepak, A.J.; Marchillo, K.; Andes, D.R. Pharmacodynamic target evaluation of a novel oral glucan synthase inhibitor, SCY-078 (MK-3118), using an in vivo murine invasive candidiasis model. Antimicrob Agents Chemother 2015, 59, 1265-1272, doi:10.1128/AAC.04445-14.
- Wring, S.A.; Randolph, R.; Park, S.; Abruzzo, G.; Chen, Q.; Flattery, A.; Garrett, G.; Peel, M.; Outcalt, R.; Powell, K., et al. Preclinical Pharmacokinetics and Pharmacodynamic Target of SCY-078, a First-in-Class Orally Active Antifungal Glucan Synthesis Inhibitor, in Murine Models of Disseminated Candidiasis. Antimicrob Agents Chemother 2017, 61, doi:10.1128/AAC.02068-16.
- Ghannoum, M.; Isham, N.; Angulo, D.; Borroto-Esoda, K.; Barat, S.; Long, L. Efficacy of Ibrexafungerp (SCY-078) against Candida auris in an In Vivo Guinea Pig Cutaneous Infection Model. Antimicrob Agents Chemother 2020, 64, doi:10.1128/AAC.00854-20.
- Vergidis, P.; Clancy, C.J.; Shields, R.K.; Park, S.Y.; Wildfeuer, B.N.; Simmons, R.L.; Nguyen, M.H. Intra-Abdominal Candidiasis: The Importance of Early Source Control and Antifungal Treatment. PLoS One 2016, 11, e0153247, doi:10.1371/journal.pone.0153247.
- Lee, A.; Prideaux, B.; Zimmerman, M.; Carter, C.; Barat, S.; Angulo, D.; Dartois, V.; Perlin, D.S.; Zhao, Y. Penetration of Ibrexafungerp (Formerly SCY-078) at the Site of Infection in an Intra-abdominal Candidiasis Mouse Model. Antimicrob Agents Chemother 2020, 64, doi:10.1128/AAC.02268-19.
- Borroto-Esoda, K.; Barat, S.; Angulo, D.; Holden, K.; Warn, P. SCY-078 Demonstrates Significant Antifungal Activity in a Murine Model of Invasive Aspergillosis. . Open Forum Infectious Diseases 2017, 4, S472., doi:https://doi.org/10.1093/ofid/ofx163.1207.
- Petraitis, V.; Petraitiene, R.; Katragkou, A.; Maung, B.B.W.; Naing, E.; Kavaliauskas, P.; Barat, S.; Borroto-Esoda, K.; Azie, N.; Angulo, D., et al. Combination Therapy with Ibrexafungerp (Formerly SCY-078), a First-in-Class Triterpenoid Inhibitor of (1-->3)-beta-d-Glucan Synthesis, and Isavuconazole for Treatment of Experimental Invasive Pulmonary Aspergillosis. Antimicrob Agents Chemother 2020, 64, doi:10.1128/AAC.02429-19.
- Barat, S.A.; Borroto-Esoda, K.; Angulo, D.; Ashbaugh, A.; Cushion, M. Efficacy of Ibrexafungerp (formerly SCY-078) in a Murine Treatment Model of PneumocystisPneumonia. ASM Microbe Online 2019.
- Spec, A.; Pullman, J.; Thompson, G.R.; Powderly, W.G.; Tobin, E.H.; Vazquez, J.; Wring, S.A.; Angulo, D.; Helou, S.; Pappas, P.G., et al. MSG-10: a Phase 2 study of oral ibrexafungerp (SCY-078) following initial echinocandin therapy in non-neutropenic patients with invasive candidiasis. J Antimicrob Chemother 2019, 74, 3056-3062, doi:10.1093/jac/dkz277.
- Wring, S.; Murphy, G.; Atiee, G.; Corr, C.; Hyman, M.; Willett, M.; Angulo, D. Lack of Impact by SCY-078, a First-in-Class Oral Fungicidal Glucan Synthase Inhibitor, on the Pharmacokinetics of Rosiglitazone, a Substrate for CYP450 2C8, Supports the Low Risk for Clinically Relevant Metabolic Drug-Drug Interactions. J Clin Pharmacol 2018, 58, 1305-1313, doi:10.1002/jcph.1146.
- Wring, S.; Murphy, G.; Atiee, G.; Corr, C.; Hyman, M.; Willett, M.; Angulo, D. Clinical Pharmacokinetics and Drug-Drug Interaction Potential for Coadministered SCY-078, an Oral Fungicidal Glucan Synthase Inhibitor, and Tacrolimus. Clin Pharmacol Drug Dev 2019, 8, 60-69, doi:10.1002/cpdd.588.
- Azie, N.; Angulo, D.; Dehn, B.; Sobel, J.D. Oral Ibrexafungerp: an investigational agent for the treatment of vulvovaginal candidiasis. Expert Opin Investig Drugs 2020, 29, 893-900, doi:10.1080/13543784.2020.1791820.
- Juneja, D.; Singh, O.; Tarai, B.; Angulo, D. Successful Treatment of Two Patients with Candida auris Candidemia with the Investigational Agent, Oral Ibrexafungerp (formerly SCY-078) from the CARES Study. Poster presentation (L0028) at 29th ECCMID congress, Amsterdam, Netherlands available online 2019.
- Cadet, R.; Tufa, M.; Angulo, D.; Nyirjesy, P. A Phase 2b, Dose-Finding Study Evaluating Oral Ibrexafungerp vs Fluconazole in Vulvovaginal Candidiasis (DOVE). Obstetrics & Gynecology 2019, 133, 113-114S, doi:10.1097/01.AOG.0000558840.33387.ee.
- Schwebke, J.R.; Sorkin-Wells, V.; Azie, N.; Angulo, D.; Sobel, J. Oral ibrexafungerp efficacy and safety in the treatment of vulvovaginal candidiasis: A phase 3, randomized, blinded, study vs. placebo (VANISH-303). IDSOG Oral Presentation 2020, 223, 964-965, doi:https://doi.org/10.1016/j.ajog.2020.08.127.
- Scynexis. SCYNEXIS Announces Positive Top-Line Results from its Second Pivotal Phase 3 Study (VANISH-306) of Oral Ibrexafungerp for the Treatment of Vulvovaginal Candidiasis (Vaginal Yeast Infection). SCYNEXIS, Inc. press release online @ https://www.globenewswire.com/news-release/2020/04/21/2019183/0/en/SCYNEXIS-Announces-Positive-Top-Line-Results-from-its-Second-Pivotal-Phase-3-Study-VANISH-306-of-Oral-Ibrexafungerp-for-the-Treatment-of-Vulvovaginal-Candidiasis-Vaginal-Yeast-Infe.html 2020.
- Scynexis. SCYNEXIS Completes Patient Enrollment Ahead of Schedule in the Second Pivotal Phase 3 Study (VANISH-306) of Oral Ibrexafungerp for the Treatment of Vulvovaginal Candidiasis (Vaginal Yeast Infection). SCYNEXIS, Inc. press release online @ https://www.scynexis.com/news-media/press-releases/detail/201/scynexis-completes-patient-enrollment-ahead-of-schedule-in 2020.
- Alexander, B.D.; Cornely, O.A.; Pappas, P.G.; Miller , R.; Johnson, M.; Vazquez, J.; Ostrosky-Zeichner, L.; Spec, A.; Rautemaa-Richardson, R.; Krause, R., et al. Efficacy and Safety of Oral Ibrexafungerpin 41 Patients with Refractory Fungal Diseases, Interim Analysis of a Phase 3 Open-label Study (FURI). Poster presented at ID week 2020 online @ https://www.scynexis.com/science/publications-and-presentations/posters-and-presentations 2019.
- Wring, S.; Borroto-Esoda, K.; Solon, E.; Angulo, D. SCY-078, a Novel Fungicidal Agent, Demonstrates Distribution to Tissues Associated with Fungal Infections during Mass Balance Studies with Intravenous and Oral [(14)C]SCY-078 in Albino and Pigmented Rats. Antimicrob Agents Chemother 2019, 63, doi:10.1128/AAC.02119-18.
- Murphy, G.; Hyman, M.; Willett, M.; Angulo, D. CYP-mediated Drug Interaction Profile of SCY-078, a novel Glucan Synthase Inhibitor (GSI). Poster presentation, ASM Microbe Online 2017.
- Lecointre, K.; Furlan, V.; Taburet, A.M. In vitro effects of tacrolimus on human cytochrome P450. Fundam Clin Pharmacol 2002, 16, 455-460, doi:10.1046/j.1472-8206.2002.00114.x.
- Scynexis. SCYNEXIS Announces FDA Acceptance and Priority Review of New Drug Application for Oral Ibrexafungerp for the Treatment of Vaginal Yeast Infections. SCYNEXIS, Inc. press release online @ https://ir.scynexis.com/press-releases/detail/225/scynexis-announces-fda-acceptance-and-priority-review-of 2020.
- Scynexis. SCYNEXIS Announces Interim Results from Phase 2 Study of Oral SCY-078 in Patients with Invasive Candidiasis. scyNEXIS, Inc. press release online @ https://www.scynexis.com/news-media/press-releases/detail/79/scynexis-announces-interim-results-from-phase-2-study-of 2016.
- Scynexis. Ibrexafungerp: First Representative of a Novel Oral/IV Antifungal Family (vF_SCYX Corporate Presentation - Dec.2020.pdf). . Corporate Presentation – Dec. 2020 online 2020.
- Kneale, M.; Bartholomew, J.S.; Davies, E.; Denning, D.W. Global access to antifungal therapy and its variable cost. J Antimicrob Chemother 2016, 71, 3599-3606, doi:10.1093/jac/dkw325.
- Mycamine (Micafungin Sodium) Injection. Fujisawa Healthcare, Inc. Application No.: 021506. Approval Date: 3/16/2005. food and Drug Administration, Center for Drug Evaluation and Research, Division of Special Pathogen and Immunologic Drug Products. [Online]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2005/21-506_Mycamine.cfm(29 December 2020, date last accessed).
- Eraxis (Anidulafungin) Injection. Vicuron Pharmaceuticals Inc. Application No.: 021632 & 021948. Approval Date: 02/17/2006. food and Drug Administration, Center for Drug Evaluation and Research, Division of Special Pathogen and Immunologic Drug Products. [Online]. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/021632_021948_EraxisTOC.cfm (29 December 2020, date last accessed).
- SAHPRA. South African Health Products Regulatory Authority Registered health products. available online @ https://www.sahpra.org.za/registered-health-products/ (accessed 17th January 2021) 2021.
This entry is adapted from the peer-reviewed paper 10.3390/jof7030163