Interferons (IFNs) are a heterogenous group of proteins that can be classified into three families (Type I, II, and III) based on distinct functions and characteristics. The family of human type I IFN is composed of 5 subgroups: IFN-α, -β, -κ, -ε, and -ω, whereas the type II IFN group only contains IFN-γ. Type III IFNs are composed of four IFN-λ proteins.
- Interferons
- innate immunity
- immune regulation
- type I interferon
1. Introduction
Type I IFNs all bind to a common heterodimeric receptor consisting of the IFN-α/β R1 (IFNAR1) and IFN-α/β R2 (IFNAR2) subunits [1][2][3], which are expressed on most cell types. Binding of type I IFNs to their receptor causes receptor subunit dimerization [4], rapid activation of the R2 subunit associated Janus kinase 1 (JAK1) [5][6], and subsequently induction of the JAK-STAT pathway [7]. This tyrosine kinase auto-phosphorylates and additionally phosphorylates specific residues within the interaction sites of the intracellular domain of the receptor, revealing signal transducer and activator of transcription (STAT) binding pockets [8]. After binding of the STAT proteins via their Src-homology 2 (SH2) domains, STATs get phosphorylated by activated JAK1, leading to their dissociation from the receptor. IFN-α induces the formation of STAT1/STAT2 heterodimers [9], which can further associate with interferon regulatory factor 9 (IRF9), and subsequently form the IFN-stimulated gene factor 3 (ISGF3) [10]. The ISGF3 translocates into the nucleus to bind interferon stimulated response elements (ISREs), inducing antiviral response genes [9][11][12]. Furthermore, STAT1 can form homodimers or heterodimers with STAT3. STAT1, STAT3, STAT4, STAT5, and STAT6 form homodimers. Dimerization precedes translocation into the nucleus and activation of genes regulated by a gamma interferon activation site (GAS) [13][14][15], causing a pro-inflammatory response (Figure 1).
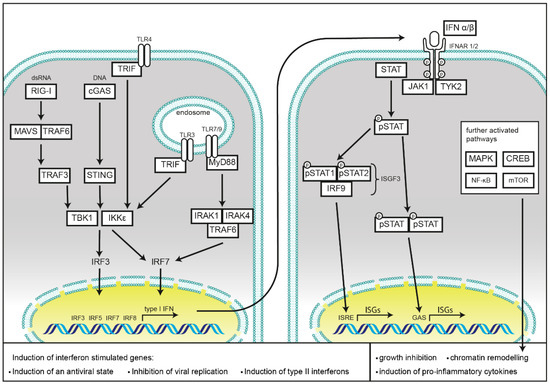
Figure 1. Schematic representation of cellular type I interferon secretion. Induction pathways and the main components of interferon production (left) and type I interferon signaling (right) are shown.
Binding of IFN-α to its receptor also leads to rapid phosphorylation of receptor subunit R1 associated tyrosine kinase Tyk2 [16][17][18][19], which mediates signaling to non-IFN pathways. This results in initiation of the MAP kinase pathway, activation of p38 and subsequent growth inhibition [20], as well as chromatin remodeling upon translocation of the Cre binding element (CREB) [21]. Furthermore, Tyk2 activates phosphoinositide-3-kinase (PI3-K), resulting in the activation of the mammalian target of rapamycin (mTOR) pathway and initiation of mRNA translation, as well as activation of the pro-inflammatory nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells (NF-κB) pathway [22].
2. Immune Response to Infection and Tissue Tolerance are Influenced by the Type I Interferon Response
Viruses interact with a broad range of proteins in mammalian cells, and their evolution has been driven by antiviral constraints and adaptation of their host cells. It is hence not surprising that their co-evolution has resulted in highly sophisticated regulatory mechanisms of the timing and amplitude of immune responses to viral challenges. Type I IFNs have a central role in controlling viral infections and are also involved in the defense of other pathogens. In 1957, IFNs were discovered by Alick Isaacs and Jean Lindenmann, as a soluble factor in the supernatant of chorio-allantoic membrane, challenged with heat inactivated influenza virus, that interferes with the viral infection in cells, hence the name “interferon” [23]. Type I IFNs act both in an autocrine and paracrine manner, and prime bystander cells for upcoming viral infection by the latter. Their ability to restrict viral replication is mainly driven by a multitude of interferon-stimulated genes (ISGs). Furthermore, type I IFNs play an important role in the activation of cells that are involved in the development of the adaptive immune response. Here type I IFNs take part in the control of cell expansion and differentiation and determining cytokine and chemokine responses of cells of the lymphoid lineage [24].
Type I IFNs are associated with the rapid induction of a cellular antiviral state, and most cells can produce them in response to an appropriate pattern-recognition receptor (PRR) stimulation. They prime the infected cells, as well as the surrounding cells towards a state of either defense or tolerance [25]. Their importance as protective factors during viral infections was proven by showing the high susceptibility of mice deficient in the IFNAR1 receptor (Ifnar1−/− mice) to vesicular stomatitis virus (VSV), Semliki Forest virus, vaccinia virus (VACV), and lymphocytic choriomeningitis virus (LCMV) [26]. Furthermore, mice with STAT1 deficiency were shown to be highly susceptible to influenza viruses [27], further cementing the importance for type I IFNs in antiviral responses. In humans, several forms of inherited STAT1 deficiencies are associated with a high susceptibility to intracellular bacteria and viruses [28], while some gain-of-function STAT1 mutations are responsible for the development of chronic mucocutaneous candidiasis [29].
In bacterial infections, the functions of type I IFNs are more complex, as they can influence host defense either positively or negatively [24]. Type I IFN treatment of macrophages results in better restriction of bacterial replication during infection with intracellular Legionella pneumophilia or Bacillus anthracis [30][31][32][33]. Furthermore, type I IFN appears to protect cells from invasion by Salmonella enterica subsp. enterica ser. Typhimurium (S. Thphimurium) and Shigella flexneri, as mice treated with recombinant type I IFN showed reduced numbers of invasive bacteria in epithelial cells and improved survival [34][35]. Type I IFNs contribute to the activation of macrophages, regarding production of nitric oxide (NO) and TNFα [36]. However, IFN-α and -β have also been identified as negative regulators of many of the cytokines and chemokines, orchestrating immune responses to bacterial infections, in particular for Listeria monocytogenes [37][38] and S. Thphimurium [38][39] (reviewed in [40]).
Besides bacteria, recognition of fungi, most importantly by the C-type lectin receptor Dectin-1, but also of fungal nucleic acids by Toll-like receptor 7 (TLR7) and TLR9 induces robust type I interferon responses [41][42]. However, as with bacterial infections, type I interferons can also be supportive for pathogen survival [43].
Type I IFNs are of equal importance in orchestrating adaptive immune responses to infection by transcriptional regulation of a broad range of target genes. Notably, type I IFNs induce and support the production of type II IFNs, mainly IFN-γ in NK cells directly [44][45], and support production of IL-12 in dendritic cells (DCs) [46]. They can further enhance responses of myeloid cells, B cells, and T cells upon viral infection, leading to improved clearance of viruses and to establishment of a robust adaptive T and B cell memory repertoire. In antigen presentation, IFN-γ induces the transcription of MHC class I and class II by inducing the expression of two NLR family members, caspase activation and recruitment domain (CARD) containing 5 (NLRC5) and MHC class II transcriptional activator (CIITA), respectively [47][48]. Meanwhile it was found that the expression of many other NLRs is regulated by both type I and type II IFNs. In the following section, we describe in detail how NLRs are regulated by type I IFNs and how they modulate the outcome of type I IFN responses. We discuss how deregulation of NLRs can result in susceptibility to either infection or auto-inflammatory disease as a consequence of pathogen dissemination or a lowered tissue tolerance to stress damage.
3. Induction of Type I Interferon Response by Nucleic Acid Sensing
Recognition of pathogen-associated molecular patterns (PAMPs) by evolutionary conserved PRRs is the initial step for mounting of a rapid innate immune response. After sensing potentially noxious non-self molecules, PRRs activate a defined set of signaling cascades, culminating in induction of a state of tolerance or defense in the host cell. This allows the production and release of cytokines, which signal to neighboring cells for recruiting immune cells for the initiation of a specific adaptive immune response.
PRRs are localized in different subcellular compartments. Toll-like receptors (TLRs), C-type lectins, and scavenger receptors cover the cell surface, as well as, in the case of TLRs, membranes of the endosomal compartment. NOD-like receptors (NLRs), RIG-I like receptors (RLRs), and cyclic GMP-AMP synthase (cGAS) monitor the cytoplasm for cell damage or the presence of invasive pathogens. Activation of these receptors results in the induction or repression of type I IFNs secretion, which will be discussed in the following chapters and is summarized in Figure 1.
Detection of cytosolic DNA is mainly mediated by the ubiquitously expressed cGAS and the absent in melanoma 2 (AIM2) protein. This not only includes foreign DNA derived from pathogens, but also cytosolic chromatin resulting from genotoxic stress. While cGAS activation induces type I IFNs, detection of cytosolic DNA by AIM2 results in pyroptotic cell death as a consequence of the activation of caspase-1 and the subsequent processing and release of mature IL-1β and IL-18 [49]. Binding to cytosolic DNA renders cGAS in an active state, leading to the synthesis of the second messenger cyclic GMP-AMP (cGAMP) with a mixed-linkage backbone (c[G(2′,5′)pA(3′,5′)p]), which in turn is sensed by the protein referred as stimulator of interferon genes (STING) [50][51][52][53], located at the membrane of the endoplasmic reticulum [54]. Activation of STING leads to its translocation into the Golgi network and activates the TRAF family member associated NF-κB activator-binding kinase 1 (TBK1). After auto-phosphorylation, TBK1 subsequently activates IRF3 through direct binding [55]. This enables its dimerization, translocation into the nucleus, and initiation of transcription of type I IFNs. IRF3 activation results in an initial wave of transcription with IFN-β and IFN-α4 as central transcription targets. Transcription of IRF7 is also induced for allowing a positive feedback loop leading to a second wave of type I IFNs secretion [56]. STING is the essential mediator of this response as its deficiency abolishes cGAS-induced IRF3 activation and IFN-β induction [57]. cGAS deficiency in mouse bone marrow-derived macrophages (BMDMs) has been shown to be detrimental to induction of antiviral type I IFN responses towards DNA viruses such as herpes simplex virus (HSV) 1, VACV, and murine gammaherpesvirus 68, but does not influence the response towards the RNA virus Sendai virus (SeV) [58][59]. Besides activation of IRF3, STING also functions as an activator of NF-κB. For an extensive review on the functions of cGAS-STING activation, the reader is referred to [60].
Studies in cells from cGAS−/− mice have proven that cGAS is the main DNA sensor in antigen presenting cells, such as plasmacytoid dendritic cells (pDCs) and conventional dendritic cells (cDCs). Depletion of cGAS in those cells rendered them unresponsive towards DNA transfection and infection with DNA viruses [61]. The type I IFN response towards these nucleic acids is also essential as a priming signal for the function of the DNA-induced AIM2 inflammasome assembly [49].
Besides nucleic acids from several DNA viruses like cytomegalovirus [62][63], HSV 1 [61], VACV [61], and retroviruses [64], cGAS is also the sensor for microbial DNA from invasive bacteria and protozoans such as L. monocytogenes [65][66][67], Chlamydia trachomatis [68], Mycobacterium tuberculosis [69][70][71], Toxoplasma gondii [72], and Leishmania major [73].
The most important family of cytosolic RNA-sensors is the RIG-I-like receptor family (RLRs), consisting of the retinoic acid-inducible gene I protein (RIG-I), melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2). These proteins are able to sense the 5-prime di- and tri-phosphates of short, blunt end double stranded (ds)RNA by RIG-I, or long dsRNA by MDA5 [74]. All three proteins contain DExD/H box domains with ATPase function, which are crucial for RNA binding. RIG-I and MDA5 further contain two CARD. These N-terminal domains are responsible for further downstream signaling by binding to the CARD domain of the mitochondrial antiviral signaling protein (MAVS). The C-terminal domain of RIG-I serves as an inhibitory domain, keeping the protein in an inactive state until it binds RNA and conformational changes are induced [75].
After the binding of different cytosolic RNA species, both MDA5 and RIG-I are subject to K63-linked ubiquitination, both by covalent, and non-covalent attachment [76]. Either RIG-I, tripartite motif-containing protein 25 (TRIM25) [76], or Riplet [77][78] can function as E3 ubiquitin ligases. This process enables RIG-I to homotetramerize [79] and localize to MAVS at the outer mitochondrial membrane initiating its oligomerization [80]. This multimerization of MAVS results in its activation and enables recruitment of additional downstream adaptor proteins TRAF2, TRAF6, and TRADD [81][82]. Subsequently TRAF3 [83] and TANK [84] are recruited to facilitate the activation of TBK1 and IKKε, which then phosphorylate the transcription factors IRF3 and IRF7. Activation of those two factors enables their homodimerization and translocation into the nucleus where they initiate transcription of type I and type III IFNs [85][86][87][88]. LGP2 does not contain a CARD domain, and hence was proposed not to function in signaling, but rather as a regulator of RIG-I or MDA5 function [89].
4. Induction of Type I Interferon Responses by Membrane Bound TLRs
While most of the members of the TLR family of TLRs may activate NF-κB signaling cascade by MyD88, type I IFNs are induced by TLRs via activation of TRIF [90]. Among those TLRs, TLR4 has proven to be the most important inducer of type I IFNs. Recognition of LPS, or several viral proteins, leads to the activation of TRIF. TRIF can then directly associate with TBK1, inducing IRF3 activation and translocation into the nucleus as described above [91][92]. Further, TLR3, which also signals via TRIF, and TLR7 and TLR9 are inducers of IFN responses [92]. TLR7 and TLR9, are mainly expressed in pDCs where they induce type I IFN expression in a MyD88-dependent manner. pDCs constitutively express IRF7, and it has been shown that MyD88 can form a complex with IRF7 to trigger its activation and transcriptional activity [93][94]. For a more comprehensive review on TLR induced immune signaling, see [95][96].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22031301
References
- Uzé, G.; Lutfalla, G.; Gresser, I. Genetic Transfer of a Functional Human Interferon Alpha Receptor into Mouse Cells: Cloning and Expression of Its Cdna. Cell 1990, 60, 225–234.
- Novick, D.; Cohen, B.; Rubinstein, M. The Human Interferon Alpha/Beta Receptor: Characterization and Molecular Cloning. Cell 1994, 77, 391–400.
- Domanski, P.; Witte, M.; Kellum, M.; Rubinstein, M.; Hackett, R.; Pitha, P.; Colamonici, O.R. Cloning and Expression of a Long Form of the Beta Subunit of the Interferon Alpha Beta Receptor That Is Required for Signaling. J. Biol. Chem. 1995, 270, 21606–21611.
- Cohen, B.; Novick, D.; Barak, S.; Rubinstein, M. Ligand-Induced Association of the Type I Interferon Receptor Components. Mol. Cell. Biol. 1995, 15, 4208–4214.
- Müller, M.; Briscoe, J.; Laxton, C.; Guschin, D.; Ziemiecki, A.; Silvennoinen, O.; Harpur, A.G.; Barbieri, G.; Witthuhn, B.A.; Schindler, C.; et al. The Protein Tyrosine Kinase Jak1 Complements Defects in Interferon-Alpha/Beta and -Gamma Signal Transduction. Nature 1993, 366, 129–135.
- Velazquez, L.; Fellous, M.; Stark, G.R.; Pellegrini, S. A Protein Tyrosine Kinase in the Interferon Alpha/Beta Signaling Pathway. Cell 1992, 70, 313–322.
- Domanski, P.; Fish, E.; Nadeau, O.W.; Witte, M.; Platanias, L.C.; Yan, H.; Krolewski, J.; Pitha, P.; Colamonici, O.R. A Region of the Beta Subunit of the Interferon Alpha Receptor Different from Box 1 Interacts with Jak1 and Is Sufficient to Activate the Jak-Stat Pathway and Induce an Antiviral State. J. Biol. Chem. 1997, 272, 26388–26393.
- Yan, H.; Krishnan, K.; Greenlund, A.C.; Gupta, S.; Lim, J.T.; Schreiber, R.D.; Schindler, C.W.; Krolewski, J.J. Phosphorylated Interferon-Alpha Receptor 1 Subunit (Ifnar1) Acts as a Docking Site for the Latent Form of the 113 Kda Stat2 Protein. EMBO J. 1996, 15, 1064–1074.
- Schindler, C.; Shuai, K.; Prezioso, V.R.; Darnell, J.E., Jr. Interferon-Dependent Tyrosine Phosphorylation of a Latent Cytoplasmic Transcription Factor. Science 1992, 257, 809–813.
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-Stat Pathways and Transcriptional Activation in Response to Ifns and Other Extracellular Signaling Proteins. Science 1994, 264, 1415–1421.
- Fu, X.Y. A Transcription Factor with Sh2 and Sh3 Domains Is Directly Activated by an Interferon Alpha-Induced Cytoplasmic Protein Tyrosine Kinase(S). Cell 1992, 70, 323–335.
- Schindler, C.; Fu, X.Y.; Improta, T.; Aebersold, R.; Darnell, J.E., Jr. Proteins of Transcription Factor Isgf-3: One Gene Encodes the 91-and 84-Kda Isgf-3 Proteins That Are Activated by Interferon Alpha. Proc. Natl. Acad. Sci. USA 1992, 89, 7836–7839.
- Ehret, B.G.; Reichenbach, P.; Schindler, U.; Horvath, C.M.; Fritz, S.; Nabholz, M.; Bucher, P. DNA Binding Specificity of Different Stat Proteins. Comparison of in Vitro Specificity with Natural Target Sites. J. Biol. Chem. 2001, 276, 6675–6688.
- Decker, T.; Lew, D.J.; Mirkovitch, J.; Darnell, J.E., Jr. Cytoplasmic Activation of Gaf, an Ifn-Gamma-Regulated DNA-Binding Factor. EMBO J. 1991, 10, 927–932.
- Levy, E.D.; Darnell, J.E., Jr. Stats: Transcriptional Control and Biological Impact. Nat. Rev. Mol. Cell. Biol. 2002, 3, 651–662.
- Barbieri, G.; Velazquez, L.; Scrobogna, M.; Fellous, M.; Pellegrini, S. Activation of the Protein Tyrosine Kinase Tyk2 by Interferon Alpha/Beta. Eur. J. Biochem. 1994, 223, 427–435.
- Colamonici, R.O.; Uyttendaele, H.; Domanski, P.; Yan, H.; Krolewski, J.J. P135tyk2, an Interferon-Alpha-Activated Tyrosine Kinase, Is Physically Associated with an Interferon-Alpha Receptor. J. Biol. Chem. 1994, 269, 3518–3522.
- Yan, H.; Krishnan, K.; Lim, J.T.; Contillo, L.G.; Krolewski, J.J. Molecular Characterization of an Alpha Interferon Receptor 1 Subunit (Ifnar1) Domain Required for Tyk2 Binding and Signal Transduction. Mol. Cell. Biol. 1996, 16, 207–282.
- Colamonici, O.; Yan, H.; Domanski, P.; Handa, R.; Smalley, D.; Mullersman, J.; Witte, M.; Krishnan, K.; Krolewski, J. Direct Binding to and Tyrosine Phosphorylation of the Alpha Subunit of the Type I Interferon Receptor by P135tyk2 Tyrosine Kinase. Mol. Cell. Biol. 1994, 14, 8133–8142.
- Caraglia, M.; Abbruzzese, A.; Leardi, A.; Pepe, S.; Budillon, A.; Baldassare, G.; Selleri, C.; de Lorenzo, S.; Fabbrocini, A.; Giuberti, G.; et al. Interferon-A Induces Apoptosis in Human Kb Cells through a Stress-Dependent Mitogen Activated Protein Kinase Pathway That Is Antagonized by Epidermal Growth Factor. Cell Death Differ. 1999, 6, 773–780.
- Lin, R.; Génin, P.; Mamane, Y.; Hiscott, J. Selective DNA Binding and Association with the Creb Binding Protein Coactivator Contribute to Differential Activation of Alpha/Beta Interferon Genes by Interferon Regulatory Factors 3 and 7. Mol. Cell. Biol. 2000, 20, 6342–6353.
- Uddin, S.; Fish, E.N.; Sher, D.A.; Gardziola, C.; White, M.F.; Platanias, L.C. Activation of the Phosphatidylinositol 3-Kinase Serine Kinase by Ifn-Alpha. J. Immunol. 1997, 158, 2390–2397.
- Isaacs, A.; Lindenmann, J. Virus Interference. I. The Interferon. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1957, 147, 258–267.
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I Interferons in Infectious Disease. Nat. Rev. Immunol. 2015, 15, 87–103.
- Stetson, D.B.; Medzhitov, R. Type I Interferons in Host Defense. Immunity 2006, 25, 373–381.
- Muller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional Role of Type I and Type Ii Interferons in Antiviral Defense. Science 1994, 264, 1918–1921.
- Koerner, I.; Kochs, G.; Kalinke, U.; Weiss, S.; Staeheli, P. Protective Role of Beta Interferon in Host Defense against Influenza a Virus. J. Virol. 2007, 81, 2025–2030.
- Boisson-Dupuis, S.; Kong, X.F.; Okada, S.; Cypowyj, S.; Puel, A.; Abel, L.; Casanova, J.L. Inborn Errors of Human Stat1: Allelic Heterogeneity Governs the Diversity of Immunological and Infectious Phenotypes. Curr. Opin. Immunol. 2012, 24, 364–378.
- Carey, B.; Lambourne, J.; Porter, S.; Hodgson, T. Chronic Mucocutaneous Candidiasis Due to Gain-of-Function Mutation in Stat1. Oral Dis. 2019, 25, 684–692.
- Plumlee, R.C.; Lee, C.; Beg, A.A.; Decker, T.; Shuman, H.A.; Schindler, C. Interferons Direct an Effective Innate Response to Legionella Pneumophila Infection. J. Biol. Chem. 2009, 284, 30058–30066.
- Schiavoni, G.; Mauri, C.; Carlei, D.; Belardelli, F.; Pastoris, M.C.; Proietti, E. Type I Ifn Protects Permissive Macrophages from Legionella Pneumophila Infection through an Ifn-Gamma-Independent Pathway. J. Immunol. 2004, 173, 1266–1275.
- Gold, A.J.; Hoshino, Y.; Hoshino, S.; Jones, M.B.; Nolan, A.; Weiden, M.D. Exogenous Gamma and Alpha/Beta Interferon Rescues Human Macrophages from Cell Death Induced by Bacillus Anthracis. Infect. Immun. 2004, 72, 1291–1297.
- Opitz, B.; Vinzing, M.; van Laak, V.; Schmeck, B.; Heine, G.; Günther, S.; Preissner, R.; Slevogt, H.; N’Guessan, P.D.; Eitel, J.; et al. Legionella Pneumophila Induces Ifnbeta in Lung Epithelial Cells Via Ips-1 and Irf3, Which Also Control Bacterial Replication. J. Biol. Chem. 2006, 281, 36173–36179.
- Bukholm, G.; Berdal, B.P.; Haug, C.; Degré, M. Mouse Fibroblast Interferon Modifies Salmonella Typhimurium Infection in Infant Mice. Infect. Immun. 1984, 45, 62–66.
- Niesel, W.D.; Hess, C.B.; Cho, Y.J.; Klimpel, K.D.; Klimpel, G.R. Natural and Recombinant Interferons Inhibit Epithelial Cell Invasion by Shigella spp. Infect. Immun. 1986, 52, 828–833.
- Mancuso, G.; Midiri, A.; Biondo, C.; Beninati, C.; Zummo, S.; Galbo, R.; Tomasello, F.; Gambuzza, M.; Macrì, G.; Ruggeri, A.; et al. Type I Ifn Signaling Is Crucial for Host Resistance against Different Species of Pathogenic Bacteria. J. Immunol. 2007, 178, 3126–3133.
- Osborne, E.S.; Sit, B.; Shaker, A.; Currie, E.; Tan, J.M.; van Rijn, J.; Higgins, D.E.; Brumell, J.H. Type I Interferon Promotes Cell-to-Cell Spread of Listeria Monocytogenes. Cell. Microbiol. 2017, 19, e12660.
- Rayamajhi, M.; Humann, J.; Penheiter, K.; Andreasen, K.; Lenz, L.L. Induction of Ifn-Alphabeta Enables Listeria Monocytogenes to Suppress Macrophage Activation by Ifn-Gamma. J. Exp. Med. 2010, 207, 327–337.
- Robinson, N.; McComb, S.; Mulligan, R.; Dudani, R.; Krishnan, L.; Sad, S. Type I Interferon Induces Necroptosis in Macrophages During Infection with Salmonella Enterica Serovar Typhimurium. Nat. Immunol. 2012, 13, 954–962.
- MacMicking, J.D. Interferon-Inducible Effector Mechanisms in Cell-Autonomous Immunity. Nat. Rev. Immunol. 2012, 12, 367–382.
- Biondo, C.; Signorino, G.; Costa, A.; Midiri, A.; Gerace, E.; Galbo, R.; Bellantoni, A.; Malara, A.; Beninati, C.; Teti, G.; et al. Recognition of Yeast Nucleic Acids Triggers a Host-Protective Type I Interferon Response. Eur. J. Immunol. 2011, 41, 1969–1979.
- del Fresno, C.; Soulat, D.; Roth, S.; Blazek, K.; Udalova, I.; Sancho, D.; Ruland, J.; Ardavin, C. Interferon-Beta Production Via Dectin-1-Syk-Irf5 Signaling in Dendritic Cells Is Crucial for Immunity to C. Albicans. Immunity 2013, 38, 1176–1186.
- Riedelberger, M.; Penninger, P.; Tscherner, M.; Seifert, M.; Jenull, S.; Brunnhofer, C.; Scheidl, B.; Tsymala, I.; Bourgeois, C.; Petryshyn, A.; et al. Type I Interferon Response Dysregulates Host Iron Homeostasis and Enhances Candida Glabrata Infection. Cell Host Microbe 2020, 27, 454–466.
- Hwang, I.; Scott, J.M.; Kakarla, T.; Duriancik, D.M.; Choi, S.; Cho, C.; Lee, T.; Park, H.; French, A.R.; Beli, E.; et al. Activation Mechanisms of Natural Killer Cells During Influenza Virus Infection. PLoS ONE 2012, 7, e51858.
- Martinez, J.; Huang, X.; Yang, Y. Direct Action of Type I Ifn on Nk Cells Is Required for Their Activation in Response to Vaccinia Viral Infection in Vivo. J. Immunol. 2008, 180, 1592–1597.
- Gautier, G.; Humbert, M.; Deauvieau, F.; Scuiller, M.; Hiscott, J.; Bates, E.E.; Trinchieri, G.; Caux, C.; Garrone, P. A Type I Interferon Autocrine-Paracrine Loop Is Involved in Toll-Like Receptor-Induced Interleukin-12p70 Secretion by Dendritic Cells. J. Exp. Med. 2005, 201, 1435–1446.
- Kobayashi, K.S.; van den Elsen, P.J. Nlrc5: A Key Regulator of Mhc Class I-Dependent Immune Responses. Nat. Rev. Immunol. 2012, 12, 813–820.
- Neerincx, A.; Castro, W.; Guarda, G.; Kufer, T.A. Nlrc5, at the Heart of Antigen Presentation. Front. Immunol. 2013, 4, 397.
- Lugrin, J.; Martinon, F. The Aim2 Inflammasome: Sensor of Pathogens and Cellular Perturbations. Immunol. Rev. 2018, 281, 99–114.
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2’,5’)Pa(3’,5’)P] Is the Metazoan Second Messenger Produced by DNA-Activated Cyclic Gmp-Amp Synthase. Cell 2013, 153, 1094–1107.
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.P.; Ludwig, J.; Hornung, V. Cgas Produces a 2’-5’-Linked Cyclic Dinucleotide Second Messenger That Activates Sting. Nature 2013, 498, 380–384.
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic Gmp-Amp Containing Mixed Phosphodiester Linkages Is an Endogenous High-Affinity Ligand for Sting. Mol. Cell 2013, 51, 226–235.
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. Sting Is a Direct Innate Immune Sensor of Cyclic Di-Gmp. Nature 2011, 478, 515–518.
- Ishikawa, H.; Barber, G.N. Sting Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678.
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural Basis of Sting Binding with and Phosphorylation by Tbk1. Nature 2019, 567, 394–398.
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Interferon [Corrected] Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360.
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic Gmp-Amp Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791.
- Li, X.; Shu, C.; Yi, G.; Chaton, C.T.; Shelton, C.L.; Diao, J.; Zuo, X.; Kao, C.C.; Herr, A.B.; Li, P. Cyclic Gmp-Amp Synthase Is Activated by Double-Stranded DNA-Induced Oligomerization. Immunity 2013, 39, 1019–1031.
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-Viral Specificity of Ifn-Induced Genes Reveals New Roles for Cgas in Innate Immunity. Nature 2014, 505, 691–695.
- Hopfner, K.P.; Hornung, V. Molecular Mechanisms and Cellular Functions of Cgas-Sting Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501.
- Li, X.D.; Wu, J.; Gao, D.; Wang, H.; Sun, L.; Chen, Z.J. Pivotal Roles of Cgas-Cgamp Signaling in Antiviral Defense and Immune Adjuvant Effects. Science 2013, 341, 1390–1394.
- Paijo, J.; Döring, M.; Spanier, J.; Grabski, E.; Nooruzzaman, M.; Schmidt, T.; Witte, G.; Messerle, M.; Hornung, V.; Kaever, V.; et al. Cgas Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLoS Pathog. 2016, 12, e1005546.
- Lio, C.W.; McDonald, B.; Takahashi, M.; Dhanwani, R.; Sharma, N.; Huang, J.; Pham, E.; Benedict, C.A.; Sharma, S. Cgas-Sting Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J. Virol. 2016, 90, 7789–7797.
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic Gmp-Amp Synthase Is an Innate Immune Sensor of Hiv and Other Retroviruses. Science 2013, 341, 903–906.
- Hansen, K.; Prabakaran, T.; Laustsen, A.; Jørgensen, S.E.; Rahbæk, S.H.; Jensen, S.B.; Nielsen, R.; Leber, J.H.; Decker, T.; Horan, K.A.; et al. Listeria Monocytogenes Induces Ifnβ Expression through an Ifi16-, Cgas- and Sting-Dependent Pathway. EMBO J. 2014, 33, 1654–1666.
- Buchmeier, N.A.; Schreiber, R.D. Requirement of Endogenous Interferon-Gamma Production for Resolution of Listeria Monocytogenes Infection. Proc. Natl. Acad. Sci. USA 1985, 82, 7404–7408.
- Handa, K.; Suzuki, R.; Matsui, H.; Shimizu, Y.; Kumagai, K. Natural Killer (Nk) Cells as a Responder to Interleukin 2 (Il 2). Ii. Il 2-Induced Interferon Gamma Production. J. Immunol. 1983, 130, 988–992.
- Zhang, Y.; Yeruva, L.; Marinov, A.; Prantner, D.; Wyrick, P.B.; Lupashin, V.; Nagarajan, U.M. The DNA Sensor, Cyclic Gmp-Amp Synthase, Is Essential for Induction of Ifn-Β During Chlamydia Trachomatis Infection. J. Immunol. 2014, 193, 2394–2404.
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The Cytosolic Sensor Cgas Detects Mycobacterium Tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 2015, 17, 811–819.
- Collins, A.C.; Cai, H.; Li, T.; Franco, L.H.; Li, X.D.; Nair, V.R.; Scharn, C.R.; Stamm, C.E.; Levine, B.; Chen, Z.J.; et al. Cyclic Gmp-Amp Synthase Is an Innate Immune DNA Sensor for Mycobacterium Tuberculosis. Cell Host Microbe 2015, 17, 820–828.
- Wassermann, R.; Gulen, M.F.; Sala, C.; Perin, S.G.; Lou, Y.; Rybniker, J.; Schmid-Burgk, J.L.; Schmidt, T.; Hornung, V.; Cole, S.T.; et al. Mycobacterium Tuberculosis Differentially Activates Cgas- and Inflammasome-Dependent Intracellular Immune Responses through Esx-1. Cell Host Microbe 2015, 17, 799–810.
- Suzuki, Y.; Orellana, M.A.; Schreiber, R.D.; Remington, J.S. Interferon-Gamma: The Major Mediator of Resistance against Toxoplasma Gondii. Science 1988, 240, 516–518.
- Green, S.J.; Crawford, R.M.; Hockmeyer, J.T.; Meltzer, M.S.; Nacy, C.A. Leishmania Major Amastigotes Initiate the L-Arginine-Dependent Killing Mechanism in Ifn-Gamma-Stimulated Macrophages by Induction of Tumor Necrosis Factor-Alpha. J. Immunol. 1990, 145, 4290–4297.
- Rehwinkel, J.; Gack, M.U. Rig-I-Like Receptors: Their Regulation and Roles in Rna Sensing. Nat. Rev. Immunol. 2020, 20, 537–551.
- Cui, S.; Eisenächer, K.; Kirchhofer, A.; Brzózka, K.; Lammens, A.; Lammens, K.; Fujita, T.; Conzelmann, K.K.; Krug, A.; Hopfner, K.P. The C-Terminal Regulatory Domain Is the Rna 5’-Triphosphate Sensor of Rig-I. Mol. Cell 2008, 29, 169–179.
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. Trim25 Ring-Finger E3 Ubiquitin Ligase Is Essential for Rig-I-Mediated Antiviral Activity. Nature 2007, 446, 916–920.
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/Rnf135, a Ring Finger Protein, Ubiquitinates Rig-I to Promote Interferon-Beta Induction During the Early Phase of Viral Infection. J. Biol. Chem. 2009, 284, 807–817.
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; Webb, A.I.; et al. Riplet, and Not Trim25, Is Required for Endogenous Rig-I-Dependent Antiviral Responses. Immunol. Cell Biol. 2019, 97, 840–852.
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural Basis for the Activation of Innate Immune Pattern-Recognition Receptor Rig-I by Viral Rna. Cell 2011, 147, 423–435.
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. Mavs Forms Functional Prion-Like Aggregates to Activate and Propagate Antiviral Innate Immune Response. Cell 2011, 146, 448–461.
- Lad, S.P.; Yang, G.; Scott, D.A.; Chao, T.H.; Jda, S.C.; de la Torre, J.C.; Li, E. Identification of Mavs Splicing Variants That Interfere with Rigi/Mavs Pathway Signaling. Mol. Immunol. 2008, 45, 2277–2287.
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. Visa Is an Adapter Protein Required for Virus-Triggered Ifn-Beta Signaling. Mol. Cell 2005, 19, 727–740.
- Saha, S.K.; Pietras, E.M.; He, J.Q.; Kang, J.R.; Liu, S.Y.; Oganesyan, G.; Shahangian, A.; Zarnegar, B.; Shiba, T.L.; Wang, Y.; et al. Regulation of Antiviral Responses by a Direct and Specific Interaction between Traf3 and Cardif. EMBO J. 2006, 25, 3257–3263.
- Guo, B.; Cheng, G. Modulation of the Interferon Antiviral Response by the Tbk1/Ikki Adaptor Protein Tank. J. Biol. Chem. 2007, 282, 11817–11826.
- Au, W.C.; Moore, P.A.; LaFleur, D.W.; Tombal, B.; Pitha, P.M. Characterization of the Interferon Regulatory Factor-7 and Its Potential Role in the Transcription Activation of Interferon a Genes. J. Biol. Chem. 1998, 273, 29210–29217.
- Au, W.C.; Moore, P.A.; Lowther, W.; Juang, Y.T.; Pitha, P.M. Identification of a Member of the Interferon Regulatory Factor Family That Binds to the Interferon-Stimulated Response Element and Activates Expression of Interferon-Induced Genes. Proc. Natl. Acad. Sci. USA 1995, 92, 11657–11661.
- Juang, Y.T.; Lowther, W.; Kellum, M.; Au, W.C.; Lin, R.; Hiscott, J.; Pitha, P.M. Primary Activation of Interferon a and Interferon B Gene Transcription by Interferon Regulatory Factor 3. Proc. Natl. Acad. Sci. USA 1998, 95, 9837–9842.
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-Dependent Phosphorylation of the Irf-3 Transcription Factor Regulates Nuclear Translocation, Transactivation Potential, and Proteasome-Mediated Degradation. Mol. Cell. Biol. 1998, 18, 2986–2996.
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and Unique Functions of the Dexd/H-Box Helicases Rig-I, Mda5, and Lgp2 in Antiviral Innate Immunity. J. Immunol. 2005, 175, 2851–2858.
- Luo, L.; Lucas, R.M.; Liu, L.; Stow, J.L. Signalling, Sorting and Scaffolding Adaptors for Toll-Like Receptors. J. Cell Sci. 2020, 133, jcs239194.
- Moynagh, P.N. Tlr Signalling and Activation of Irfs: Revisiting Old Friends from the Nf-Kappab Pathway. Trends Immunol. 2005, 26, 469–476.
- Uematsu, S.; Akira, S. Toll-Like Receptors and Type I Interferons. J. Biol. Chem. 2007, 282, 15319–15323.
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.; Uematsu, S.; et al. Interferon-Alpha Induction through Toll-Like Receptors Involves a Direct Interaction of Irf7 with Myd88 and Traf6. Nat. Immunol. 2004, 5, 1061–1068.
- Honda, K.; Yanai, H.; Mizutani, T.; Negishi, H.; Shimada, N.; Suzuki, N.; Ohba, Y.; Takaoka, A.; Yeh, W.C.; Taniguchi, T. Role of a Transductional-Transcriptional Processor Complex Involving Myd88 and Irf-7 in Toll-Like Receptor Signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 15416–15421.
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and Localization of Toll-Like Receptor Signalling Complexes. Nat. Rev. Immunol. 2014, 14, 546–558.
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat. Immunol. 2010, 11, 373–384.