Gastric cancer (GC) is ranked third in cancer deaths world-wide. It is separated anatomically into either gastric adenocarcinomas (non-cardia GC) or gastro-esophageal-junction adenocarcinomas (cardia GC) and is further classified histologically into either diffuse or intestinal types.
- metabolomics
- gastric cancer
- esophageal cancer
- colorectal cancer
- Warburg effect
- metabolome
1. Introduction to Gastric Cancer
Gastric cancer (GC) is ranked third in cancer deaths world-wide. It is separated anatomically into either gastric adenocarcinomas (non-cardia GC) or gastro-esophageal-junction adenocarcinomas (cardia GC) and is further classified histologically into either diffuse or intestinal types [1][2]. Genomic profiling of primary non-cardia GC has led to the identification of four tumor subgroups: 9% positive for Epstein-Barr virus, 22% microsatellite unstable, 20% genomically stable and 50% chromosomally unstable tumors [1]. Each of these four types of tumors have specific histology and gene mutations associated with them (Figure 1A) [1]. Helicobacter pylori infection is a major cause of sporadic GC due to the contribution of the chronic inflammation induced by its colonization of the stomach [3]. True hereditary GC accounts for about 1–3% of all GC mainly due to mutations in Cadherin-1 (CDH1) and catenin alpha-1 (CTNNA1) mutations [4]. Environmental factors that are thought to promote GC include, low consumption of fruits and vegetables, high intake of salt, nitrates and pickled foods, smoking, obesity and gastro-esophageal-reflux (GERD) disease [5][6][7].
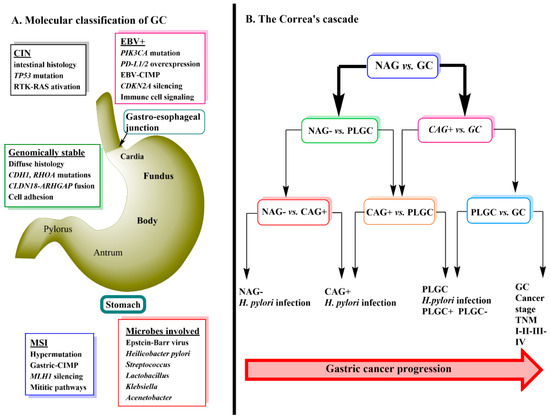
GC progression is often described by a sequence of events known as Correa’s Cascade (Figure 1B) [12]. The cascade begins with non-active gastritis (H. pylori negative) (NAG-) which then proceeds to chronic active gastritis (H. pylori positive) (CAG+) → precursor lesions of GC (PLGC) (atrophy, intestinal metaplasia, dysplasia) → GC [12][13]. The prognosis of GC is related to the stage at which it is diagnosed with the treatment being surgical removal of the cancer tissue and resection. Currently, diagnosis relies heavily on invasive techniques such as endoscopy with biopsy followed by pathological examination [14]. Early stages of GC are often asymptomatic [15] and therefore, the use of metabolomics to detect serum biomarkers that would allow an earlier diagnosis of GC has become an important area of ongoing research.
2. The Metabolomics Profile of Gastric Cancer
In this section, we will begin by examining recent GC cancer metabolomics papers and extracting data regarding the most significantly different metabolites between GC and healthy controls found in blood. Table 1 summarizes three individual studies plus the findings of a meta-analysis for nine GC studies in blood [2][14][16][17]. Metabolomics studies have also been performed in urine [18] but as we are comparing three different cancers (GC, EC, CRC), only one sample type (blood) was used for the comparison. The main reason for choosing blood over urine samples is that 24 h urine samples need to be collected in order to get accurate metabolite detection and quantification. This is often inconvenient for the patient making complete compliance more difficult, whereas a fasting blood sample is much easier to obtain. The goal here was to examine potential metabolite profiles that could discriminate between the three cancers based on findings from recent studies.
Table 1. Description of patient cohorts and study types used in GC studies.
| Reference [19] | Reference [14] | Reference [2] | Reference [17] |
|---|---|---|---|
| 184 GC/208 HC Unmatched Case control study Untargeted metabolomics |
20 GC/19 HC Unmatched 68/43 GC/HC mean age 8 F/12 M (GC) Targeted metabolomics panel of 216 metabolites |
84 GC/82 non-GC Unmatched 28–79 age GC 25–82 age non-GC 45 M/39 F (GC) Targeted metabolomics panel of Amino acids |
104 GC/50 HC Unmatched Untargeted metabolomics |
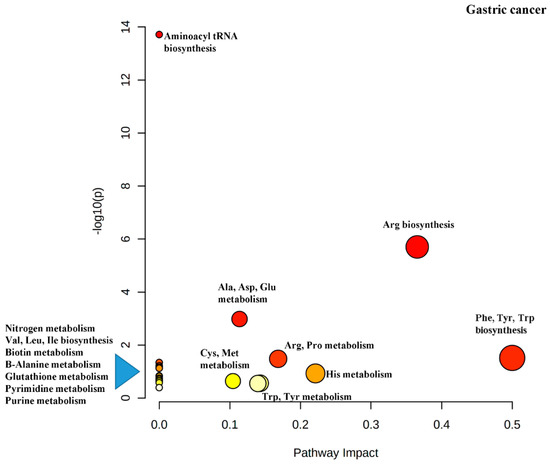
Another output from the MetaboAnalyst software is the impact map for the various metabolic pathways resulting from the pathway enrichment analysis (Figure 4). It can be seen that Phe, Tyr and Trp biosynthesis has been rated as important (indicated by dark red color), has several differential metabolites in the pathway (indicated by the size of the circle) and has the greatest impact (indicated by the x-axis).
The metaboloites listed in Table 4 are highyly relevant to cancer metabolism.It is well-known that oxidative phosphorylation of glucose is impaired in the mitochondria of cancer cells as a result of the Warburg effect and thus, the number of the acetyl-CoA molecules derived from glucose is significantly reduced. Instead, cancer cells rely on upregulation of amino acid biosynthesis and metabolism to replenish TCA cycle intermediates to generate ATP [2][20] Trp catabolism is important for the production of Acetyl-CoA for use in the TCA cycle and both His and Trp are used for the anabolism of one-carbon units for the synthesis of nucleotides for DNA and RNA biosynthesis [21]. Kynurenine, a major metabolite of Trp via the enzyme indoleamine-2,3-dioxygenase (IDO1), induces immunosuppression by binding to and activating the transcription factor aryl hydrocarbon receptor (AhR) [22][23]. This inhibits the ability of immune-tolerant dendritic cells (DCs) and regulatory T cells to target and eliminate cancer cells [24][25].
Glu is also used by cancer cells in the TCA cycle and it can be biosynthesized by the transamination of His and Arg and/or by the deamination of glutamine [20][26]. Thr catabolism via Thr dehydrogenase produces Gly and acetyl-CoA which can also feed the TCA cycle [27]. Met can also enter the Met-folate cycle to produce 1-carbon units for nucleobases [28]. All of the abovementioned amino acids were found to be reduced in the serum of GC patients possibly due to the upregulation of these metabolic pathways in the tumor. The Arg-Ornithine-polyamine pathway may be used to synthesize polyamines that promote the proliferation of cancer cells. Ornithine can be converted to citrulline in the urea cycle to replenish Arg supplies as Arg is decreased in most tumors [26][29][30]. Tyr is important for integration into proteins that activate important oncogenic signaling pathways such as Kras [31].
There is much interest in targeting amino acid metabolism to treat cancer. The alanine-serine-cysteine transporters 2 (ASCT2, encoded by SLC1A5 has been spotlighted as a therapeutic target because it is the primary glutamine transporter [26]. Gln is the amino acid that is consumed the most by cancer cells and inhibition of glutaminase which converts Gln to Glu, by CB-839 is in pre-clinical and clinical trials [32][33]. The enzyme glutamate dehydrogenase is responsible for the bioconversion of Glu to α-ketoglutamate for use in the TCA cycle and inhibition of this enzyme has been used to inhibit tumor growth [34]. Asparaginase, an injectable enzymatic drug that degrades asparagine in the plasma and is used as a treatment for acute lymphoblastic leukemia. The lack of Asn in cells cause apoptosis [35] Kinase inhibitors such as, imatinib and dasatinib, which act on receptors that bind Tyr containing proteins have been used successfully for treatment of GI cancer [36]. IDO1 inhibitors are actively being evaluated in clinical trials [28] The IDO1/Kynurinine/AhR pathway is also being investigated for its therapeutic potential [37]. Figure 4 is a diagram of the pathway impact for the discriminating metabolites just discussed.
In summary, although differences in study design and assay conditions resulted in many non-reproducible differential metabolites between the various studies there were still some which were reported consistently. Pathway enrichment analysis was performed on those metabolites and their significance to cancer and some therapeutic opportunities were discussed. In the next section we are going to use the same approach to examine the metabolomics of EC.
3. The Role of Gut Microbiota Produced Metabolites in GI Cancer
Increases in certain microbial metabolites also play a role in GI cancer. As we have just pointed out for CRC, increased amounts of lactate derived from lactic acid producing bacteria are able to cause upregulation of the MCT1 transporter to allow increased amounts of lactate into the cancer cell where it acts a tumor promoting substance [38]. Lactate-derived pyruvate can stabilize hypoxia-inducible factor-1 (HIF-1) by inhibiting HIF polyhydroxylases which in tumor endothelial cells, stimulates angiogeneisis [39]. Lactate also mediates M2-like polarization of tumor associated macrophages contributing to immunosuppression and immune escape of cancer cells [40].
While butyrate has been shown to have beneficial effects, in the context of APC mutations as is found in nearly all of CRC, butyrate was shown to promote proliferation of aberrant epithelial cells contributing to increased cancer polyp formation [41]. Acetaldehyde, especially in saliva is mainly of microbial source and contributes to upper GI cancer by causing molecular damage and mutagenesis [42]. The esophageal microbiome is a reflection of the oral microbiome [43] and oral pathogens have been implicated in CRC as well [44].
Secondary and tertiary bile acids (BAs) are bacterial metabolites that have been implicated in GI carcinogenesis [45]. In a recent study of human BA reflux gastritis, it was found that in patients with BA reflux gastritis that there were higher amounts of conjugated primary and secondary BAs, notably, glycocholic acid (GCA), glycochenodeoxycholic acid (GCDCA), glycodeoxycholic acid (GDCA), taurodeoxycholic acid (TDCA), taurocholic acid (TCA) and taurochenodeoxycholic acid (TCDCA) in their gastric juice In contrast, normal and gastritis patients without BA reflux had equal amounts of conjugated and unconjugated BAs in their gastric juice [46].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13040720
References
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric cancer. Lancet 2016, 388, 2654–2664.
- Jing, F.; Hu, X.; Cao, Y.; Xu, M.; Wang, Y.; Jing, Y.; Hu, X.; Gao, Y.; Zhu, Z. Discriminating gastric cancer and gastric ulcer using human plasma amino acid metabolic profile. IUBMB Life 2018, 70, 553–562.
- Bornschein, J.; Selgrad, M.; Warnecke, M.; Kuester, D.; Wex, T.; Malfertheiner, P.H. pylori infection is a key risk factor for proximal gastric cancer. Dig. Dis. Sci. 2010, 55, 3124–3131.
- Oliveira, C.; Pinheiro, H.; Figueiredo, J.; Seruca, R.; Carneiro, F. Familial gastric cancer: Genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015, 16, e60–e70.
- Lunet, N.; Valbuena, C.; Vieira, A.L.; Lopes, C.; Lopes, C.; David, L.; Carneiro, F.; Barros, H. Fruit and vegetable consumption and gastric cancer by location and histological type: Case-control and meta-analysis. Eur. J. Cancer Prev. 2007, 16, 312–327.
- Ladeiras-Lopes, R.; Pereira, A.K.; Nogueira, A.; Pinheiro-Torres, T.; Pinto, I.; Santos-Pereira, R.; Lunet, N. Smoking and gastric cancer: Systematic review and meta-analysis of cohort studies. Cancer Causes Control. 2008, 19, 689–701.
- Yang, P.; Zhou, Y.; Chen, B.; Wan, H.W.; Jia, G.Q.; Bai, H.L.; Wu, X.T. Overweight, obesity and gastric cancer risk: Results from a meta-analysis of cohort studies. Eur. J. Cancer 2009, 45, 2867–2873.
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209.
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649.
- Fleisher, A.S.; Esteller, M.; Wang, S.; Tamura, G.; Suzuki, H.; Yin, J.; Zou, T.T.; Abraham, J.M.; Kong, D.; Smolinski, K.N.; et al. Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res. 1999, 59, 1090–1095.
- Meng, C.; Bai, C.; Brown, T.D.; Hood, L.E.; Tian, Q. Human Gut Microbiota and Gastrointestinal Cancer. Genom. Proteom. Bioinform. 2018, 16, 33–49.
- Kuligowski, J.; Sanjuan-Herraez, D.; Vazquez-Sanchez, M.A.; Brunet-Vega, A.; Pericay, C.; Ramirez-Lazaro, M.J.; Lario, S.; Gombau, L.; Junquera, F.; Calvet, X.; et al. Metabolomic Analysis of Gastric Cancer Progression within the Correa’s Cascade Using Ultraperformance Liquid Chromatography-Mass Spectrometry. J. Proteome Res. 2016, 15, 2729–2738.
- Wang, D.; Li, W.; Zou, Q.; Yin, L.; Du, Y.; Gu, J.; Suo, J. Serum metabolomic profiling of human gastric cancer and its relationship with the prognosis. Oncotarget 2017, 8, 110000–110015.
- Lario, S.; Ramirez-Lazaro, M.J.; Sanjuan-Herraez, D.; Brunet-Vega, A.; Pericay, C.; Gombau, L.; Junquera, F.; Quintas, G.; Calvet, X. Plasma sample based analysis of gastric cancer progression using targeted metabolomics. Sci. Rep. 2017, 7, 17774.
- Park, Y.H.; Kim, N. Review of atrophic gastritis and intestinal metaplasia as a premalignant lesion of gastric cancer. J. Cancer Prev. 2015, 20, 25–40.
- Tian, J.; Xue, W.; Yin, H.; Zhang, N.; Zhou, J.; Long, Z.; Wu, C.; Liang, Z.; Xie, K.; Li, S.; et al. Differential Metabolic Alterations and Biomarkers Between Gastric Cancer and Colorectal Cancer: A Systematic Review and Meta-Analysis. OncoTargets Ther. 2020, 13, 6093–6108.
- Huang, S.; Guo, Y.; Li, Z.; Zhang, Y.; Zhou, T.; You, W.; Pan, K.; Li, W. A systematic review of metabolomic profiling of gastric cancer and esophageal cancer. Cancer Biol. Med. 2020, 17, 181–198.
- Chen, Y.; Zhang, J.; Guo, L.; Liu, L.; Wen, J.; Xu, L.; Yan, M.; Li, Z.; Zhang, X.; Nan, P.; et al. A characteristic biosignature for discrimination of gastric cancer from healthy population by high throughput GC-MS analysis. Oncotarget 2016, 7, 87496–87510.
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484.
- Wang, H.; Zhang, H.; Deng, P.; Liu, C.; Li, D.; Jie, H.; Zhang, H.; Zhou, Z.; Zhao, Y.L. Tissue metabolic profiling of human gastric cancer assessed by (1)H NMR. BMC Cancer 2016, 16, 371.
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637.
- Greene, L.I.; Bruno, T.C.; Christenson, J.L.; D’Alessandro, A.; Culp-Hill, R.; Torkko, K.; Borges, V.F.; Slansky, J.E.; Richer, J.K. A Role for Tryptophan-2,3-dioxygenase in CD8 T-cell Suppression and Evidence of Tryptophan Catabolism in Breast Cancer Patient Plasma. Mol. Cancer Res. 2019, 17, 131–139.
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.L.; Puccetti, P. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 2003, 4, 1206–1212.
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089.
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198.
- Choi, B.H.; Coloff, J.L. The Diverse Functions of Non-Essential Amino Acids in Cancer. Cancers 2019, 11, 675.
- Bastings, J.; van Eijk, H.M.; Olde Damink, S.W.; Rensen, S.S. d-amino Acids in Health and Disease: A Focus on Cancer. Nutrients 2019, 11, 2205.
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30.
- Hu, L.; Gao, Y.; Cao, Y.; Zhang, Y.; Xu, M.; Wang, Y.; Jing, Y.; Guo, S.; Jing, F.; Hu, X.; et al. Identification of arginine and its “Downstream” molecules as potential markers of breast cancer. IUBMB Life 2016, 68, 817–822.
- Shu, X.L.; Xu, H.; Yu, T.T.; Zhong, J.X.; Lei, T. Regulation of apoptosis in human gastric cancer cell line SGC-7901 by L-arginine. Panminerva Med. 2014, 56, 227–231.
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359.
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219.
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901.
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 2014, 56, 414–424.
- Marini, B.L.; Perissinotti, A.J.; Bixby, D.L.; Brown, J.; Burke, P.W. Catalyzing improvements in ALL therapy with asparaginase. Blood Rev. 2017, 31, 328–338.
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48.
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325.
- Nasr, R.; Shamseddine, A.; Mukherji, D.; Nassar, F.; Temraz, S. The Crosstalk between Microbiome and Immune Response in Gastric Cancer. Int. J. Mol. Sci. 2020, 21, 6586.
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418.
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563.
- Bultman, S.J.; Jobin, C. Microbial-derived butyrate: An oncometabolite or tumor-suppressive metabolite? Cell Host Microbe 2014, 16, 143–145.
- Nieminen, M.T.; Salaspuro, M. Local Acetaldehyde-An Essential Role in Alcohol-Related Upper Gastrointestinal Tract Carcinogenesis. Cancers 2018, 10, 11.
- Norder Grusell, E.; Dahlen, G.; Ruth, M.; Ny, L.; Quiding-Jarbrink, M.; Bergquist, H.; Bove, M. Bacterial flora of the human oral cavity, and the upper and lower esophagus. Dis. Esophagus 2013, 26, 84–90.
- Zhang, Y.; Yu, X.; Yu, E.; Wang, N.; Cai, Q.; Shuai, Q.; Yan, F.; Jiang, L.; Wang, H.; Liu, J.; et al. Changes in gut microbiota and plasma inflammatory factors across the stages of colorectal tumorigenesis: A case-control study. BMC Microbiol. 2018, 18, 92.
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214.
- Zhao, A.; Wang, S.; Chen, W.; Zheng, X.; Huang, F.; Han, X.; Ge, K.; Rajani, C.; Huang, Y.; Yu, H.; et al. Increased levels of conjugated bile acids are associated with human bile reflux gastritis. Sci. Rep. 2020, 10, 11601.