Hepatocellular carcinoma (HCC) is the seventh most frequent cancer and the fourth leading cause of cancer mortality worldwide. MicroRNAs (miRNAs) are small, endogenous, noncoding, single-stranded RNAs that modulate the expression of their target genes at the posttranscriptional and translational levels. Aberrant expression of miRNAs has frequently been detected in cancer-associated genomic regions or fragile sites in various human cancers and has been observed in both HCC cells and tissues. The precise patterns of aberrant miRNA expression differ depending on disease etiology, including various causes of hepatocarcinogenesis, such as viral hepatitis, alcoholic liver disease, or nonalcoholic steatohepatitis.
- microRNA
- hepatocellular carcinoma
- pathogenesis
- biomarker
1. Introduction
Liver cancer is the sixth most frequent malignancy and the fourth leading cause of cancer-related deaths worldwide [1]. Hepatocellular carcinoma (HCC) accounts for approximately 80% of all liver cancers and is a main cause of cancer mortality [2]. The risk factors of hepatocarcinogenesis include various liver diseases, such as infections caused by hepatitis B virus (HBV) and hepatitis C virus (HCV), alcoholic liver disease (ALD), nonalcoholic steatohepatitis (NASH), and nonalcoholic fatty liver disease (NAFLD) [3,4,5]. Despite recent great progress in HCC therapy, the 5-year survival rate for advanced-stage HCC remains poor owing to its late diagnosis, resistance to anticancer therapy, and high frequency of recurrence [6,7,8]. Therefore, elucidating the detailed underlying mechanisms and pathogenesis of HCC is important for the development of new diagnostic and prognostic biomarkers and therapeutic drugs.
MicroRNAs (miRNAs, miRs) are small, endogenous, interfering, noncoding RNAs of 21–30 nucleotides in length. More than 2600 miRNAs have been predicted to be encoded by the human genome, with the ability to modulate more than 15,000 genes [9]. Each miRNA negatively regulates target genes by binding to the 3′ untranslated region (UTR) of mRNAs. These complexes are involved in RNA-mediated interference, and in vertebrates, mRNA transcripts are usually not cleaved by a miRNA-associated RNA-induced silencing complex (RISC) but rather undergo translational repression and degradation via deadenylation [10]. The miRNA-associated RISC can suppress gene expression [11,12]. Remarkably, a single miRNA can modulate more than 200 mRNAs [13,14].
The biogenesis of miRNAs includes several steps, such as transcription, cleavage, export, further cleavage, strand selection, and interaction with mRNAs [15] (Figure 1). Generally, RNA polymerase II transcribes primary miRNA (pri-miRNAs) transcripts in the canonical pathway or mirtron pathway [16]. The 5′ ends of pri-miRNAs are capped, and the 3′ ends are polyadenylated [17]. Pri-miRNAs are then cleaved to 70–100-nucleotide hairpin-structured precursors (pre-miRNAs) by a nuclear RNase III enzyme named Drosha, which is a double-stranded RNA-binding domain protein involving DiGeorge syndrome critical region 8 (DGCR8)/Pasha as a cofactor [18]. Next, pre-miRNAs are exported from the nucleus to the cytoplasm, after binding with exportin-5 and Ran-GTP [19] and are cleaved by Dicer [20], with transactivation response element RNA-binding 70 protein (TRBP) serving as a cofactor for Dicer [20]. Finally, double-stranded pre-miRNAs undergo rapid unwinding, when loaded onto Argonaute (AGO), and only one strand, which serves as a guide to target mRNAs, remains bound [21]. These RISCs with the AGO protein modulate mRNA degradation and translational inhibition [21].
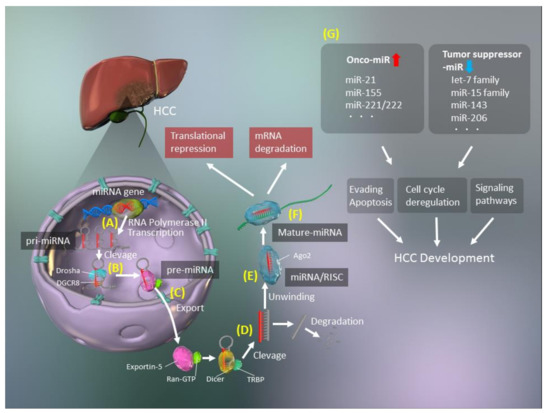
However, the biogenesis of miRNAs is more diverse and may involve noncanonical pathways that bypass Drosha/DGCR8 processing. There are several noncanonical types of miRNAs, including mirtrons, small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs), and short hairpin RNAs (shRNAs) [22]. Mirtrons are pre-miRNAs that are generated without Drosha by pre-mRNA splicing and intron debranching [23,24]. SnoRNAs [25] and tRNAs [26,27] have different internal hairpin structures, and their processing involves the cleavage activity of Dicer, without Drosha/DGCR8. Transcripts of shRNA, which are derived from unannotated and intergenic regions, can function as Dicer substrates during transcription [28,29].
Although the important roles of miRNAs in the modulation of mRNA expression are well established, their precise functions remain elusive. Interestingly, miRNAs modulate the expression of approximately 30% of all human genes, many of which are tumor-associated or are in regions of instability in the genome [30,31]. There is clear evidence of key roles for miRNAs in human carcinogenesis [13,32,33,34,35], with two types of miRNAs identified, namely, oncogenic miRNAs (oncomiRs) and tumor suppressor miRNAs (Figure 1). OncomiRs induce carcinogenesis by inhibiting the expression of tumor suppressors, while tumor suppressor miRNAs inhibit oncogene expression in normal cells and are lacking in cancer cells. Two miRNAs, miR-15 and miR-16, were first reported to be altered in cancer and are associated with a frequently targeted chromosomal deletion of BCL2, which encodes an anti-apoptotic factor [36].
Several reports have demonstrated relationships between miRNAs and HCC [35,37,38,39,40] and identified potential miRNA biomarkers and therapeutic targets for HCC diagnosis and treatment.
2. Roles of miRNAs in the Liver
2.1. Lipid Metabolism
The liver plays a critical role in lipid metabolism. Dysfunctions in lipid metabolism induce excessive accumulation of hepatic triglycerides and fatty acids, resulting in various liver diseases, such as NAFLD and NASH. Several miRNAs have pivotal functions in the maintenance of cholesterol and fatty acid metabolism [41], and others, such as miR-33, miR-103, miR-104, and miR-307, act as modulators of lipid and cholesterol levels [42].
Various serum miRNAs, including miR-122, miR-21, miR-34a, and miR-451, are enhanced in patients with NAFLD [43]. A representative liver-specific miRNA, miR-122, which is highly upregulated in the liver, is involved in hepatic cholesterol and lipid metabolism in this disease [44]. Suppression of miR-122 can diminish the plasma cholesterol levels, reduce hepatic fatty acid and cholesterol synthesis, and increase the oxidation of hepatic fatty acids [45]. In addition, miR-122 is associated with hepatic lipogenesis-related enzymes, including fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC1). The inhibition of miR-122 also decreases hepatic lipogenesis in obese mice [44].
Several reports have demonstrated that miR-34a is associated with hepatic steatosis [46,47]. This miRNA inhibits hepatic silent information regulator 1 (sirtuin 1, SIRT1), peroxisome proliferator-activated receptor-α (PPARα), and liver X receptor (LXR) [46,47,48]. Reduced SIRT1 levels in the liver of patients with NAFLD can be recovered by the inhibition of miR-34a, which results in the improvement of hepatic steatosis via PPARα and the activation of AMP-activated protein kinase (AMPK) [47].
2.2. Glucose Metabolism
miR-375, a novel islet-specific miRNA, can inhibit glucose-induced insulin secretion, whereas inhibition of miR-375 can increase insulin secretion. This miR-375-associated insulin modulation is independent of altered glucose metabolism and is related to insulin exocytosis. Myotrophin (MTPN), which is a target of miR-375, is also involved in glucose metabolism. The inhibition of MTPN enhances glucose-induced insulin secretion and insulin exocytosis, indicating that miR-375 might be a new therapeutic target for diabetes mellitus and NAFLD [49]. Expression of miR-23a is increased in a NASH-related HCC murine model, and overexpression of miR-23a via the interleukin-6 (IL-6)/STAT3 signaling pathway decreases glucose production through the inhibition of PGC1α and G6PC expression [50]. In addition, miR-143, which is involved in insulin resistance, controls the ORP8-dependent regulatory pathway of AKT, and miR-143 overexpression reduces insulin-stimulated AKT activation [51]. Moreover, miR-206 decreases lipid and glucose levels in hepatocytes by modulating lipogenesis and insulin signaling [52], which suggests that miR-206 might be a diagnostic biomarker and therapeutic target for NAFLD and hyperglycemia.
2.3. Hepatic Inflammation
Hepatic inflammation, which involves inflammatory cytokine production and endoplasmic reticulum stress, results from an abnormal immune response; the latter is mediated by several factors, such as viral and bacterial infections, metabolic disorders, alcohol abuse, drug allergies, and toxic reagents [53]. Various miRNAs, such as miR-122 and miR-132, play pivotal roles in the innate and adaptive immunity involved in hepatic inflammation. miR-122 is the most representative miRNA in the liver, and its loss results in inflammation, fibrosis, and HCC, indicating that miR-122 modulates anti-inflammatory effects [54,55]. Furthermore, miR-122 may inhibit hepatic infiltration of inflammatory cells and the secretion of various cytokines, including IL-6 and tumor necrosis factor-α (TNFα), by these cells [54,55]. miR-132, which inhibits the expression of SIRT1, is a mediator of inflammation in chronic liver diseases. The overexpression of miR-132 induces the translocation of nuclear factor-κB (NF-κB) into the nucleus, acetylation of p65, and production of IL-8 and MCP-1. The loss of miR-132, which is mediated by serum deprivation, diminishes the acetylation level of p65 and partially downregulates the expression of IL-8 and MCP-1. Therefore, the inhibition of miR-132 has anti-inflammatory effects in the liver.
2.4. Hepatic Fibrosis
Fibrosis is developed in the liver as a result of continuous and severe hepatocyte damage, resulting in an inflammatory cytokine storm. Several miRNAs can synergistically modulate inflammatory signaling pathways. Furthermore, several miRNAs are associated with the activation of hepatic stellate cells (HSCs) and progression of hepatic fibrosis via the regulation of related signaling pathways.
Members of the miR-29 family induce apoptosis via the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway and modulate the accumulation of the extracellular matrix (ECM) [56,57,58]. Stimulation of HSCs by transforming growth factor-beta (TGF-β) promotes myofibroblastic transition and ECM induction, resulting in liver fibrogenesis. TGF-β1 mediates the downregulation of miR-29 in HSCs [57]. In addition, miR-29 overexpression in mouse HSCs leads to the reduction of collagen-1α1 and collagen-4α1 levels [57,59,60] via modulation of various ECM genes. Similar to other hepatic fibrosis-related miRNAs, members of the miR-34 family promote hepatic fibrosis by activating HSCs, whereas those of the miR-378 family inhibit the development of fibrosis in a GLIS-dependent manner. The miR-15 family is associated with the induction of cell proliferation and apoptosis, and its members regulate the TGF-β signaling pathway by inhibiting TGFβR1, SMAD3, SMAD7, p38, and endoglin in cardiac fibrosis [61]. The miR-199 and miR-200 families are involved in the secretion of profibrotic cytokines and are responsible for ECM deposition [62,63]. These miRNA families act as modulators of hepatic fibrosis by targeting genes involved in fibrosis-related signaling pathways and activating HSCs.
3. Roles of miRNAs in Various Liver Diseases Leading to HCC
3.1. HBV Infection
HBV infection is one of the most prevalent risk factors of HCC development [64]. Thus, HBV-related HCC is a serious health concern worldwide [64]. Recently, many reports have demonstrated that miRNAs play pivotal roles in each stage of HBV-related HCC development [65,66,67,68]. HBV dysregulates miRNAs that modulate the expression of the host/HBV genes during HCC pathogenesis. In fact, this type of HCC is characterized by a range of immune response failures due to the dysregulation of miRNAs [69].
The HBV X protein (HBx) inhibits miR-34 expression via p53 stimulation in hepatocytes, which results in the upregulation of a macrophage-derived chemokine (CCL22), stimulation of regulatory T cells (Tregs), and the suppression of effector T cells, thereby increasing HBV genome transcription [70,71]. HBx-induced upregulation of miR-155 leads to a reduction in the suppressor of cytokine signaling-1 (SOCS1) expression, increasing JAK/STAT signaling and suppressing HBV infection mediated by the induction of interferon (IFN) signaling [72]. In addition, HBx-induced miR-155 blocks CCAAT/enhancer-binding protein (C/EBP), which activates the HBV enhancer (Enh) 11/core promoter and then inhibits HBV replication [73].
Liver-specific miR-122 is upregulated in the serum after HBV infection and is regarded as one of the key modulators of HBV replication [74,75,76]. However, Wang et al. [77] demonstrated that HBx suppressed miR-122 expression and increased the levels of HBV transcripts by blocking p53 binding to the HBV Enh1/core promoter. Another report has revealed that miR-122 blocks HBV pregenome RNA, which encodes the hepatitis B core antigen and viral polymerase and inhibits HBV replication via dysregulation of heme oxygenase-1 (HO-1), which suppresses refill of HBV covalently closed circular DNA [78]. In addition, upregulation of several miRNAs, including miR-184, miR-185, miR-196a, miR-199a-3p, miR-210, and miR-217, directly affects HBV transcription [79]. Furthermore, it has recently been demonstrated that the HBV virion produces its own HBV-miR-2 and HBV-miR-3 [80]. HBV-miR-3 inhibits mRNA expression of the hepatitis B core (HBc) protein, which is involved in HBV self-regulation [81], to prolong survival by escaping the host immune system [79,82]. miR-372 also modulates HBV expression, which depends on target pathways. HBx-upregulated miR-372 [83,84] targets the cyclic AMP response element-binding protein (CREB) by binding to HBV Enh1/core promoter (ENI-Cp), thereby suppressing HBV transcription. miR-372 targets nuclear factor 1 B-type protein (NFIB), which modulates HBV Enh1/ENI-Cp, thus increasing HBV transcript levels [79,83]. Various miRNAs are thus related to the modulation of HBV transcript levels and the host immune response to the virus.
3.2. HCV Infection
HCV contributes to serious liver problems, such as liver cancer, including HCC [85]. IFN-free, direct-acting antiviral therapy improves HCV treatment, with sustained virologic response rates greater than 95%, without many side effects [86]. However, the high mutation rate of HCV may result in the resistance to direct-acting antivirals, and patients with mutated HCV have low sustained virologic response rates [87]. Therefore, further studies of the molecular mechanism underlying HCV infections are needed to improve HCV therapy.
Dysregulation of miRNAs due to HCV infection occurs via multiple pathways, such as the immune response, lipid metabolism, and cell-cycle pathways [88]. Several target genes, such as PPARG and fibronectin 1 (FN1), were found to be downregulated, while other target genes, including stearoyl-CoA desaturase (SCD) and CREB1, were found to be upregulated by at least 11 miRNAs, namely, miR-130a/b, miR-200, miR-34a, miR-23b, miR-24, miR-146a, miR-381, miR-25, miR-200a, and miR-371-5p, after HCV infection [89]. In addition, miR-122 modulates the host immune response by regulating the expression of target genes and directly targeting the HCV genome [90,91,92]. In particular, miR-122 stabilizes the 5′ and 3′ UTRs of the HCV genome, and inhibition of this miRNA dramatically reduces the replication of HCV RNA [93,94]. By contrast, miR-141, miR-192, miR-215, and miR-491 increase HCV replication. Thus, miR-141 enhances HCV replication by reducing the tumor suppressor deleted in liver cancer 1 (DLC-1) [95], and miR-491 enables HCV entry via the PI3K/AKT signaling pathway [96]. However, miR-196, miR-29, let-7b, miR-130a, and miR-27a induce anti-HCV activity [59,97,98,99,100,101]. It has also been found that miR-199a suppresses HCV replication by blocking domain II of the internal ribosomal entry site [97]. In addition, IFN-β enhances the expression of several miRNAs with anti-HCV activity, such as miR-196, miR-431, and miR-448. Among these, miR-196 and miR-448 may directly target the HCV RNA genome [91]. These findings suggest that several miRNAs are strongly involved in the modulation of HCV infection and replication.
3.3. ALD
ALD is a complex disease caused by prolonged and heavy alcohol consumption, along with predisposing genetic factors. ALD can cause liver dysfunction, including steatohepatitis, fibrosis, cirrhosis, and eventually HCC [102]. There is tremendous evidence of the effects of miRNA dysregulation on the pathogenesis and development of ALD, such as liver damage, lipid metabolism dysfunction, inflammation, oxidative stress, apoptosis, and fibrosis [102]. Inflammation-related miRNAs, such as miR-132, miR-155, miR-146, and miR-21, influence alcohol/lipopolysaccharide (LPS)/TLR4 pathways. TLR4 transmits proinflammatory stimuli via a mitogen-activated protein kinases (MAPKs) or TIR domain-containing adaptor-inducing IFN-β (TRIF) [103]. The expression of miR-212 is increased by alcohol in intestinal cells, and LPS is also induced by alcohol in the liver via the suppression of ZO-1 [104,105]. Alcohol-induced oxidative stress downregulates miR-199a in liver sinusoidal endothelial cells by upregulating ET1 and hypoxia-inducible factor 1α (HIF1α), which are related to steatohepatitis and fibrosis in the liver [106]. The downregulation of miR-223 increases neutrophil infiltration in the liver and induces liver injury by inhibiting the IL-6/p47phox/reactive oxygen species axis [107]. The enhanced expression of miR-217 exacerbates alcoholic fatty changes by damaging SIRT1 and lipin-1 [108]. Moreover, upregulation of miR-34a in ALD results in the development of liver fibrosis via caspase-2, SIRT1, and matrix metallopeptidase (MMP) 1 and MMP2 [109]. In addition, miR-122 modulates hepatic lipid metabolism and inflammation [110]. The miRNA let-7 is diminished in response to alcohol and loss of let-7 induces a mesenchymal phenotype in HSCs to enhance liver injury by inhibiting LIN28B. Finally, an imbalance between let-7 and LIN28/28B causes oncogenesis [111].
3.4. NAFLD and NASH
NAFLD is defined as steatosis in at least 5% of hepatocytes [112], without other liver diseases, including viral hepatitis and autoimmune, alcohol-related, and genetic liver diseases. Recently, NAFLD has become one of the most frequent liver diseases worldwide [113]. Various miRNAs have been identified to be involved in NASH development. For example, the levels of miR-122, which is a representative hepatic miRNA, are 7.2 times higher in patients with NASH than in healthy subjects and 3.1 times higher than in patients with simple steatosis [114]. Liver-specific miR-122-knockout mice rapidly develop NASH and exhibit enhanced lipogenesis, changes in lipid secretion, IL-6 and TNF-α production, and upregulation of chemokine (C–C motif) ligand 2 (CCL2). Decreased miR-122 expression enhances fibrogenesis by inducing HIF1α and MAPK1, which can also facilitate HCC development [115]. In addition, miR-192 is involved in the development of TGF-β1-promoted fibrosis, which activates SMAD signaling [116]. However, members of a miRNA superfamily that includes miR-16, miR-497, miR-195, miR-322, and miR-15, particularly miR-15 and miR-16, regulate hepatic fibrosis and hepatocarcinogenesis [61]. The development and progression of NASH increases the risk of HCC via miRNAs. A recent report has demonstrated that miRNAs are pivotal for the activation of HSCs during NASH development [117]. Free cholesterol is accumulated because of increases in both SREBP2 and miR-33a signaling via the inhibition of PPARγ signaling, along with HSC activation and disruption of the SREBP2-induced cholesterol feedback system [117]. Upregulation of miR-21, which downregulates the expression of the tumor suppressor phosphatase and tensin homolog (PTEN), is mediated by unsaturated fatty acids in hepatocytes [118]. Furthermore, the expression of miR-155, which inhibits another tumor suppressor gene, C/EBPβ, is enhanced in mice fed a choline-deficient, amino acid-defined diet [73,119].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13030514