E. coli is a versatile microorganism, and even if the invasive infections are those that are more likely to evolve with life threatening complications, the diarrheagenic strains of E. coli are still important pathogens, especially in the pediatric departments.
- diarrheal disease
- typical/atypical enteropathogenic E. coli
- virulence factors
- diagnostic
1. Introduction
Diarrheal disease is still a major health problem, and it is considered an important cause of morbidity and mortality, especially in infants and children under five years old. Although the disease primarily affects populations from developing countries, where sanitation, water supplies, and the medical addressability rate are inadequate, there are still reported cases in developed countries [1,2,3,4]. The etiology of the infectious diarrheal disease is influenced by the geographic area. Viruses (such as rotavirus, adenovirus, norovirus) are the most frequent etiological agents worldwide. Infections with Campylobacter spp. and Salmonella spp. are frequently reported in the developed countries, while diarrheagenic strains of E. coli, Vibrio spp. are encountered mostly in developing countries [5,6,7].
In 1885, Theodor Escherich, a German pediatrician described, for the first time, Bacterium coli commune, as a commensal Gram-negative rod from the healthy individual’s intestinal flora. These rods were afterward renamed Escherichia coli (E. coli), in his honor. Only after a few decades, in 1935, they were considered pathogens, being associated with an outbreak of “cholera infantum”, a severe gastroenteritis affecting neonates [8,9]. From that moment, a remarkable number of studies were conducted, describing E. coli from numerous points of view (serological, antibiotic resistance, molecular, genetic engineering, sequencing, etc.), leading to the fact that, in our days, it is considered as one of the best-characterized bacteria. Even so, there are still many unknowns about this pathogen, which, even nowadays, is responsible for a large number of infections in healthy or immunocompromised persons (diarrhea, pneumonia, urinary, wounds infections, sepsis, meningitidis) [10,11].
E. coli is a versatile microorganism, and even if the invasive infections are those that are more likely to evolve with life threatening complications, the diarrheagenic strains of E. coli are still important pathogens, especially in the pediatric departments. Among these strains, Enteropathogenic E. coli (EPEC) is a particularly important one, as it is a bacterial etiological agent in a pathology that is dominated by viruses, making it easy to be “forgotten” or not taken into consideration, especially in the case of infantile diarrhea. The fact that these strains present multiple phenotypic differences, that they possess different, multiple virulence factors (some of them belonging to other pathotypes), and that they are still evolving, makes them a group of bacteria that will always be of interest for physicians from different medical departments. The main objective of this review was to summarize the most important, relevant, and up to date information available in the literature about this reemerging pathogen, and to provide an overview of the most important characteristics of EPEC strains.
The diarrheagenic strains of E. coli are a very homogeneous group of intestinal pathogens, with different characteristics regarding the antigen structure, virulence mechanisms, host colonization sites, and clinical evolution [11]. The O (lipopolysaccharide) and H (flagellar) antigens are used for the serological classification of the pathogenic strain of E. coli. Because of the high diversity of these antigens (187 O antigens, 53 H antigens), currently, serotyping is considered a laborious, costly, and unreliable diagnostic tool due to the cross-reactivity between different serogroups [12,13]. Most of the recent diagnostic protocols recommend the classification/identification of diarrheagenic E. coli using molecular methods, which can identify the virulence factors of these strains. Based on the presence/absence of virulence factors, the diarrheagenic strains of E. coli are classified into 6 major pathotypes and a hybrid one (Table 1). The horizontal gene transfer between different strains is an important mechanism that leads to the diversity of these strains, keeping them in a constant movement between the classically described pathotypes. Some strains that combine virulence factors from different pathotypes are considered hybrids, and they can be potentially more virulent than the original strains [6,11,14]. Plasmids, bacteriophages, transposons, pathogenicity islands, insertion sequences (mobile genetic elements–MGEs) plays important roles in the genomic plasticity of E. coli [15]. Whole genome sequencing is considered the gold standard technique which can accurately serotype strains and evaluate relatedness among isolates based on differences in gene content or allelic variation [16,17,18,19]. This technique revealed that E. coli presents a conserved core of genes (common to pathogenic and commensal strains as well) that acquired, by horizontal gene transfer, small cluster of genes and genomic islands associated with virulence [20].
Table 1. Pathotypes of Diarrheagenic E. coli.
| Pathotype of Diarrheagenic E. coli | Acronym | Virulence Factors | Diagnostic Targets for PCR | Disease | Clinical Symptoms | Ref |
|---|---|---|---|---|---|---|
| Enteropathogenic E. coli |
EPEC | Locus of enterocyte effacement (LEE), intimin, bundle-forming pilus | bae, bfpA | Acute/persistent diarrhea in children | Watery diarrhea, vomiting | [6,17,21] |
| Enterohaemorrhagic E. coli (Shiga toxin-producing) |
EHEC/STEC | Shiga toxin 1 and/or 2 LEE, adhesins (EHEC) |
stx1, stx2, eae, ehxA, bfp | Hemolytic-uremic syndrome, hemorrhagic colitis | Bloody diarrhea | [6,11,17] |
| Enteroinvasive E. coli |
EIEC | Shiga toxin, hemolysin, cellular invasion, Ipa | ipaH, other ipa genes | Shigellosis-like syndrome | Watery, bloody diarrhea | [6,11] |
| Adherent Invasive E. coli |
AIEC | Type 1 fimbriae, cellular invasion | none | Associated with Crohn disease | Persistent intestinal inflammation | [12,17,22] |
| Enterotoxigenic E. coli |
ETEC | Heat-labile and heat-stable toxins, CFAs (colonization factors) | elt, est | Traveler’s diarrhea | Watery diarrhea, vomiting | [6,11,17] |
| Diffusely Adherent E. coli |
DAEC | Adhesins | Afa/Dr adhesins | Diarrhea in children | Watery diarrhea | [6,17,23] |
| Enteroaggregative E. coli (hybrid pathotype) |
EAEC | Adhesins, toxins and secreted proteins | pAA, AggR, AAFs | Diarrhea in children | Vomiting diarrhea (with mucus) | [6,11] |
2. EPEC Definition and Classification
EPEC was the first identified pathotype of diarrheagenic E. coli, being the causative agent of a few outbreaks of infantile diarrhea in 1940–1950 [24]. Until a few decades ago, the O (somatic), H (flagellar), and K (capsular) antigens were the basis for the classification of EPEC strains. In 1987, when World Health Organization recommended that O26, O55, O86, O111, O114, O119, O125, O126, O127, O128, O142, and O158 should be considered as EPEC serogroups, some studies already reported that some of these serogroups included strains from other serotypes [14,25]. Serogroups such as O39, O88, O103, O145, O157, and O158 are now considered to be included in EPEC pathotype [26]. Among EPEC isolates, H2 and H6 are the most common flagellar antigens associated, but there are also some less common H types (such as H7, H8, H9, H12, H21, H27, H25, and H34), and some EPEC strains are classified as H negative (non-motile) [11,14,24,25,27]. Because of the high diversity of these antigens, serotyping is no longer considered a rapid diagnostic tool, nor a reliable one [13,28,29].
In 1982, EPEC was defined as “diarrheagenic E. coli belonging to serogroups epidemiologically incriminated as pathogens but whose pathogenic mechanisms have not been proven to be related either to heat-labile enterotoxins or heat-stable enterotoxins or to Shigella-like invasiveness” [30]. Now, EPEC strains are defined, based on their virulence factors, as diarrheagenic strains of E. coli that can produce attaching and effacing (A/E) lesions on the intestinal epithelium, but unable to produce Shiga toxins and heat-labile (LT) or heat-stable (ST) enterotoxins [3,11,24].
The most important characteristic of EPEC is the ability to produce A/E lesions, which enable them to cause localized lesions by attaching tightly to the surface of the intestinal epithelial cells, disrupting the cell surfaces, finally leading to the effacement of the microvilli. Intimin is an outer membrane protein, encoded by the eae gene, which mediates the intestinal cell attachment. The attachment is facilitated by Tir (Translocated intimin receptor), an effector inserted in the host plasma membrane, where it functions as a receptor for intimin. By a type-three-secretion-system (T3SS), EPEC can inject a large number (at least twenty-five) effector proteins in the host cells. All the required genetic elements for the A/E lesion are encoded on the locus of enterocyte effacement (LEE), a large genomic pathogenicity island [31,32,33,34].
EPEC can also be classified on typical and atypical isolates, based on the presence or absence of EAF (plasmid E. coli adherence factor), where bfp and per, two important operons, are localized. bfp encodes the type IV bundle-forming pilus (BFP), while per encodes the plasmid-encoded regulator (Per), a transcriptional activator [31]. All the EPEC strains are eae positive (+), so typical EPEC are described as eae+, bfpA+, while atypical strains are considered those eae+, bfpA− (negative) [11,27,31,35]. Some of the atypical EPEC strains might possess other virulence factors such as enteroaggregative heat-stable toxin (EAST1), hemolysin, not encoded on LEE [26].
3. Virulence Factors
EPEC can adhere, at least in vitro, to cell lines and organ cultures in a three-dimensional microcolonies pattern, named localized adherence (LA) pattern. BFP mediates LA phenotype, and it is also involved in antigenicity, biofilm formation, and autoaggregation [11]. EPEC are non-invasive pathogens that can induce lesions by introducing effector proteins directly into the host cells, using a type-three-secretion-system (T3SS). These effectors are encoded on the LEE pathogenicity island. LEE contains genes that encode T3SS (Esc, Sep), the outer membrane adhesin (intimin, essential for the adherence on the host cells, encoded by eae gene), translocators (EspA/B/D), effector proteins (EspF/G/H, Map, EspZ), chaperones (Ces proteins), the translocated intimin receptor (Tir), and regulatory proteins such as LEE-encoded regulator (Ler), a global regulator of LEE proteins (GrlA—activator, GrlR—repressor) [11,36,37,38,39,40].
Various non-LEE (Nle) encodes effector genes (cif, espI/nleA, nleB, nleC, nleD, nleE, nleH), located outside of the LEE region, clustered in six pathogenicity islands. The Nle proteins can increase the bacterial virulence by preventing/modulating the host inflammatory response and by disrupting the tight junctions and the cytoskeleton of the host cells [11,31,33,36]. Along with Nle A, EspF, and Map, other non-LEE encoded effector proteins are involved in the disassembly of the tight junctions [41]
In addition to BFP, some EPEC strains may present other fimbriae or pili (type 1 fimbriae, E. coli common pilus), flagella, astA gene (encoding EAST1—the enteroaggregative E. coli heat-stable enterotoxin 1), autotransporter proteins (EspC), or produce a hybrid adherence phenotype in HeLa cells (LA and aggregative-like pattern), but their involvement in the pathogenicity still needs to be studied [11,42].
4. Pathogenesis
Attaching-and-effacing was described by Nataro and Kaper, in 1998, as the “hallmark” of the EPEC infections, a lesion that is characterized by the intimate attachment of the bacteria to the host epithelial cell membrane and the effacement of the microvilli [25]. Since then, a lot of studies have focused on describing this main pathogenic mechanism, which is common to the typical and atypical EPEC strains. The interaction between EPEC and the host cells is described as a four-stage process (Figure 1).
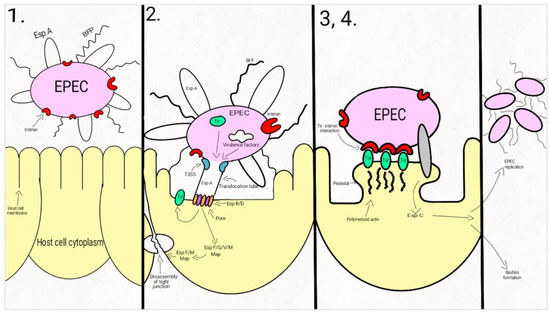
Figure 1. Schematic representation of the EPEC adherence mechanism: 1. EPEC express Bfp and EspA; 2. After attaching to the enterocyte (trough Bfp), EPEC use a T3SS to infect many effectors in the cell. The phosphorylated Tir is inserted into the host cell membrane; 3. The bacterial intimin binds to the modified Tir, attaching the bacteria to the host cell. Actin and cytoskeletal elements are accumulated near the site of the bacterial adherence. EspC is inserted into the cell trough an autotransporter system, T5SS. 4. The cytoskeletal elements, accumulated near the site of the attachment, leads to the formation of the pedestal structure, characteristic for EPEC. In the first step, EPEC cells express the intimate adhesin intimin, Bfp (bundle-forming pili), and EspA (short filaments surface-associated). Environmental factors regulate the expression of these virulence factors, influencing the site of the bacterial colonization (small/large bowel) [43,44,45].
In the second step, EPEC strains adhere to the intestinal epithelium through Bfp and EspA, forming dense microcolonies on the cell’s surface, in a pattern described as localized adherence. The type-three-secretion-system creates a pore, enabling the bacteria to inject Tir and a large number (at least 25, up to 50) of effector molecules into the host cell. These effectors facilitate bacterial colonization, immune evasion, and regulate inflammatory response and host cell death. They also activate the host cell-signaling pathways, causing the alteration of the cytoskeleton, leading to the loss of the microvilli. After tyrosine-protein kinase and protein kinase A modifies Tir, it is inserted into the host cell membrane [43,44,46,47,48,49,50,51,52,53,54].
Enterocyte effacement and intimate bacterial attachment to the host cell characterizes the third step of EPEC infection. The bacterial cells lose the EspA filaments from their surface. The bacterial intimin (encoded by eae) binds to the modified Tir, causing the intimate attachment of the bacteria to the host cell. In this phase, actin and cytoskeletal elements are accumulated near the site of the bacterial adherence [43,44,55]. An autotransporter system, T5SS (type V secretion system), mediates the secretion of EspC, [42,56,57,58], a protein involved not only in epithelial cell cytotoxicity, but also in bacterial replication and biofilm formation [56,59,60,61].
During the fourth step, the cytoskeletal elements, accumulated near the site of the attachment, leads to the formation of the pedestal structure, characteristic for EPEC. The effector molecules (translocated from the bacteria) disrupt the cell processes, leading, eventually, to cell death [24,43,62].
The clinical symptoms of EPEC diarrheal disease occurs before the complete establishment of the A/E lesions and the loss of the microvilli, so these mechanisms are most likely involved in the exacerbation of the diarrhea. The rapid onset of the diarrhea (sometimes only after a few hours after the ingestion) is more likely to be the result of a secretory mechanism [63,64].
EPEC can imbalance the host cells electrolyte transport, and by the dysregulation of these transport pathways, it is responsible for the rapid onset of the symptoms. Na+/H+ (NHE2-3), Na+/glucose (SGLT-1), and Cl−/HCO3− (DRA/PAT1) exchangers are responsible for the water and solutes intestinal absorption. CFTR (the apical cAMP-dependent cystic fibrosis transmembrane conductance regulator) is responsible, among other factors, for the osmotic gradient which controls the movement of water into the intestinal lumen. EPEC establish a disequilibrium in the Na+/Cl− electroneutral exchange through the plasma membrane, reducing water absorption. Moreover, by modulating the AQP (epithelial aquaporin) expression, the water transport is directly altered by EPEC [63,65,66,67,68].
EPEC is capable of disrupting the epithelial barrier function and structure. By activating MLC kinase, EspB-mediated phosphorylation, activation of PKCα, EPEC can increase the paracellular permeability. EPEC is also responsible for the disruption of epithelial apical junctional complexes and the alteration of the tight junctional proteins [63].
The inflammatory response is considered more likely to be involved in the severity and the duration of the disease than in the early stages of the infection. An increase of IL-1β, TNFα, interferon (IFN) γ in the infected mucosa is associated with the EPEC infection. Both pro-inflammatory and anti-inflammatory pathways of the epithelial cells are involved in the EPEC mediated inflammatory response. The EPEC secreted components are responsible for the pro-inflammatory response, while the effectors inserted in the host cells by the T3SS attenuates the inflammatory response [49,63,69].
This entry is adapted from the peer-reviewed paper 10.3390/gastroent12010004