Klotho was initially introduced as an antiaging molecule. Klotho deficiency significantly reduces lifespan, and its overexpression extends it and protects against various pathological phenotypes, especially renal disease. It was shown to regulate phosphate and calcium metabolism, protect against oxidative stress, downregulate apoptosis, and have antiinflammatory and antifibrotic properties.
- Klotho protein
- diabetes mellitus
- diabetic nephropathy
1. Introduction
α-Klotho was discovered in 1997 and simply named Klotho because no other Klotho proteins were identified at that time. It was shown to be an antiaging molecule. Mice that exhibited Klotho deficiency presented a premature aging phenotype, whereas Klotho overexpression extended their lifespan by up to 30% and protected them against many pathological phenotypes, especially renal disease [1][2].
Klotho is expressed mostly in the kidneys and choroid plexus in the brain. Its expression was also detected in the pituitary, parathyroid glands, the heart, and reproductive organs, with low levels in other tissues [1]. In the kidneys, Klotho is present in podocytes, the (mostly) apical and basolateral membrane, and intracellularly in cells that comprise the proximal tubule of the nephron. Klotho is also shed to the proximal tubule lumen[3][4]. Kidney cells are the main source of soluble Klotho, but it can also be shed from ependymal cells of the choroid plexus[5]. Soluble Klotho was detected in blood, urine, and cerebrospinal fluid and is considered to be a hormone that exerts beneficial systemic effects[1]. The reason why soluble Klotho can target so many tissues and affect various signaling pathways is currently unknown because no specific receptor for soluble Klotho has yet been identified. The pleiotropic effects of soluble Klotho are hypothesized to be based on its capacity to bind sialic acid and target monogangliosides (e.g., monosialotetrahexosylganglioside—GM1 and monosialodihexosylganglioside—GM3) that are enriched in lipid rafts of cell membranes. Soluble Klotho can also interact with numerous intracellular proteins[5].
2. Structure and Function of Klotho
Two or three forms of α-Klotho protein exist. Membrane-bound Klotho is a 130 kDa single-pass transmembrane protein that is encoded by a KL gene. The extracellular domain of the Klotho protein can be cleaved and separated from the cell membrane by the metalloproteinases ADAM-10 and ADAM-17 (ADAM—a disintegrin and metalloproteinase domain-containing protein) and β-secretase 1 (BACE1). This proteolytic cleavage by ADAM-10 and ADAM-17 can be stimulated inter alia by insulin. The extracellular domain of Klotho contains two homologous repeat sequences (KL1 and KL2), which are separated from each other by unknown proteases. Both the whole released extracellular domain and separated KL1 and KL2 are referred to as soluble Klotho particles[1]. Another, alternative transcript of the KL gene that possesses a STOP codon was thought to result in the production of a shorter, secretory form of the protein[6]. However, according to some researchers, this transcript is degraded and not translated into a protein product. Moreover, splicing of the two (standard functional and alternative) mRNA transcripts of Klotho in humans is dysregulated especially in acute kidney injury (AKI), favoring the nonfunctional alternative transcript. This might contribute to the decrease in the expression of functional Klotho protein also in diabetic nephropathy[2].
Both transmembrane Klotho and soluble Klotho serve as coreceptor proteins for fibroblast growth factor (FGF) 23 (binding to FGF receptors 1–4), which promotes phosphaturia to control phosphate metabolism[2]. Klotho also functions independently to regulate many signaling pathways because it is evolutionarily older than FGF23 [7]. Klotho regulates not only phosphate metabolism but also calcium metabolism. Furthermore, it inhibits the insulin/insulin-like growth factor 1 (IGF-1) signaling pathway and activates forkhead box (FoxO) transcription factors, resulting in the production of antioxidant enzymes (e.g., catalase and manganese-dependent superoxide dismutase [8]) and reduction of oxidative stress through the removal of ROS, thereby downregulating apoptosis. It also suppresses tumor necrosis factor α (TNF-α)-induced oxidative damage and prevents the translocation of nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB), a transcription factor of many proinflammatory genes, including TNF-α, to the nucleus. Klotho also suppresses the profibrotic transforming growth factor β1 (TGF-β1) and Wnt/β-catenin signaling[9][1].
3. Role of Klotho in Diabetes and Diabetic Nephropathy
The disruption of phosphate and calcium metabolism, oxidative stress, inflammation, the fibrotic process, and an increase in the ratio of β-cell and podocyte loss through apoptosis can result in pathologies that resemble premature aging. Klotho protects cells against accelerated aging and damage during the course of diabetes mellitus (DM) and diabetic nephropathy (DN) [1][6]. Moreover, a reduction of plasma and urine levels of circulating soluble Klotho was observed during the aging process in cells with short telomeres or stress-induced premature senescence, widespread tissue injury, inflammation, oxidative stress and vascular calcification, which occur during the course of chronic kidney disease (CKD). Additionally, in patients with CKD, low levels of calcitriol (i.e., the bioactive form of vitamin D3) were found to intensify renal Klotho deficiency [9]. Notably, the leading cause of CKD in Western countries is diabetic nephropathy [21]. Low levels of Klotho mRNA and protein were also expected to be observed in diabetic nephropathy. This assumption was confirmed in diabetic mice. In two models of T1DM, Klotho deficiency promoted the apoptosis of insulin-producing β-cells, which were protected against this process after Klotho overexpression. Furthermore, Klotho improved the β-cell function and prevented the development of type 2 DM. Low plasma, but not urine, levels of Klotho predicted the progression of nephropathy in T2DM patients and were negatively correlated with a decrease in the glomerular filtration rate (GFR)[1][10]. Moreover, diabetes-induced proteinuria, oxidative stress (reflected by intracellular ROS levels), podocyte injury, and apoptosis that was caused by protein kinase Cα (PKCα) activation were aggravated by Klotho deficiency and partially ameliorated by Klotho overexpression[11]. Both plasma and urine levels of soluble Klotho were lower in intrinsic AKI patients compared with prerenal AKI patients, together with an increase in levels of proinflammatory ligands (i.e., S100A8/A9 calgranulins) of Toll-like receptor 4 (TLR4) [12]. Therefore, plasma or urine levels of the Klotho protein were proposed to serve as a biomarker of early kidney injury in diabetic patients.
4. Immune Response in Diabetes and Diabetic Nephropathy
Type 1 DM is triggered by unknown environmental factors and develops in individuals with a polygenetic predisposition through the destruction of insulin-producing pancreatic β-cells. The damage to these cells is caused by impairments in the immune response and local inflammation (Figure 1), including the infiltration of pancreatic islets by M1 macrophages, T and B lymphocytes (CD8+ and CD4+ T cells and CD20+ B cells), and the expression of proinflammatory cytokines, especially interleukin 1β (IL-1β) and TNF-α. High levels of proinflammatory and profibrotic IL-6 are produced in initial stages of T1DM. Moreover, IL-6-inducible autoimmunity-related gene (HIP/PAP) has been shown to be expressed in the pancreas in patients with T1DM, providing further evidence that IL-6 participates in the autoimmune process in type 1 DM[13].
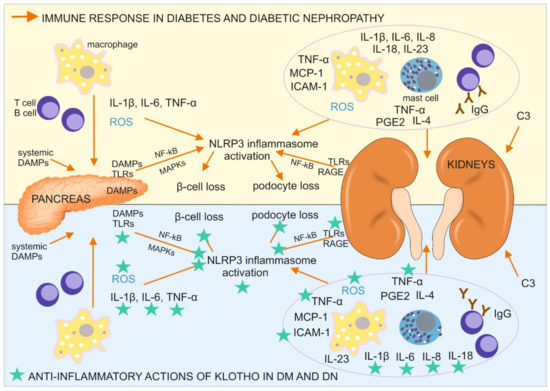
Figure 1. Immune response in diabetes mellitus (DM) and diabetic nephropathy (DN) with a designation of its tissue location, together with antiinflammatory actions of Klotho, directed on the cells, pathways, and proteins involved in the described immune response. C3, complement component 3; DAMPs, danger associated molecular patterns; ICAM-1, intercellular adhesion molecule 1; IgG, immunoglobulin G; IL, interleukin; MAPKs, mitogen-activated protein kinases; MCP-1, monocyte chemoattractant protein 1; NF-κB, nuclear factor κ-light-chain-enhancer of activated B cells; NLRP3 inflammasome, a type of inflammasome that contains NACHT, LRR, and PYD domain-containing protein 3; PGE2, prostaglandin E2; RAGE, receptor for advanced glycation endproducts; ROS, reactive oxygen species; TLR, Toll-like receptor; TNF-α, tumor necrosis factor α.
Type 2 DM develops during the course of a prolonged high-glucose diet and consists of impairments in pancreatic insulin secretion and a lower cellular response to insulin stimulation. These metabolic changes are accompanied by a low-grade inflammatory process in adipose tissue, the liver, and pancreatic islets, in which IL-1β plays a major role[14]. The prolonged upregulation of IL-1β leads to an increase in serum insulin levels, which promotes glucose uptake by macrophages, which infiltrate the pancreatic tissue, with an upregulation of their proinflammatory activity, including the production of ROS and initiation of the generation of the NLRP3 (NOD-, LRR-, and pyrin domain-containing protein 3) inflammasome[15]. The resulting inflammation in the pancreas leads to β-cell loss, whereas the immune reaction in adipocytes and liver cells, especially IL-1β–NLRP3 inflammasome pathway activation, results in a lower insulin response[16]. NLRP3 inflammasome activation in the pancreas in diabetic patients is instigated by oxidative stress and systemic, and islet tissue-derived danger associated molecular patterns (DAMPs), including high-mobility group box 1 (HMGB1), heat shock protein 70 kDa (HSP70), islet amyloid polypeptide, and fatty acids (e.g., palmitate). These DAMPs can bind TLR2 and TLR4, which are expressed by cells in pancreatic islets and pancreatic macrophages, trigger the activation of NF-κB and mitogen-activated protein kinases (MAPKs), and further promote NLRP3 inflammasome activation. Another DAMP, thioredoxin-interacting protein (TXNIP), was shown to initiate posttranslational activation of the NLRP3 inflammasome in T2DM and trigger IL-1β production, resulting in the inflammation of pancreatic islets and apoptosis of β cells[15]. Another interleukin, IL-6, was shown to participate in the development of type 2 DM. IL-6 levels were higher in patients with T2DM, which exacerbated insulin resistance and caused atherosclerosis through its role in accelerating inflammation[13].
This multiorgan inflammatory reaction, accompanied by higher levels of its marker C-reactive protein (CRP), causes pathological changes in the microvascular circulation in the kidneys, macrophage infiltration of this organ, and the local production of a wide range of proinflammatory cytokines and mediators that are detectable beginning in early stages of diabetic nephropathy in serum and peripheral blood cells in affected individuals. The inflammatory response in DM is accelerated by the formation and accumulation of advanced glycation end products (AGEs), which are bind by Immunoglobulins G (Ig G), produced by B cells. AGE-Ig G immune complex can be then bound to receptors for Fc fragment of Ig G (FcγRs), phagocyted and presented on the surface of antigen presenting cells (also podocytes) to T lymphocytes[16].
Other receptors also contribute to an accelerated immune response, albuminuria, and podocyte damage in diabetic nephropathy. These are intracellular nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs; e.g., NOD2) that initiate NLRP3 inflammasome formation and the expression of proinflammatory molecules by immune cells (e.g., IL-1β, IL-6, IL-8, IL-18, TNF-α, and monocyte chemoattractant protein 1 (MCP-1), also called chemokine-CC motif ligand (CCL2)) and intracellular adhesion molecule 1 (ICAM-1)[16]. Notably, some of these receptors (e.g., TLR4, RAGE, some FcRs, and NOD2) are not only expressed on/in immune cells but also on/in podocytes (continuously or upon stimulation) where they activate antigen (e.g., AGEs) phagocytosis, antigen presentation with major histocompatibility complex (MHC) molecules to T cells, and inflammasome formation, leading to apoptotic podocyte loss[17][18]. The production of proinflammatory molecules can also occur in mesangial cells, which possess receptors for Fc fragments of immunoglobulin G (FcγRs), which are crucial for antigen recognition and engulfment during phagocytosis. Mice with FcγR deficiency were protected against inflammation, glomerular damage, and albuminuria in diabetic nephropathy[16]. Furthermore, the levels of other inflammation-associated receptors (e.g., TLR2, TLR4, and RAGE) were elevated in diabetic nephropathy kidney tissue, together with DAMPs, such as AGEs, HMGB1, HSP70, and S100 calgranulins (e.g., S100A8 and S100A9), triggering their activation. The expression and function of TLR4 also increased in monocytes in diabetes patients[19]. Signaling through these TLRs is associated with myeloid differentiation factor 88 (MyD88) protein stimulation. Signaling through TLR4 and RAGE leads to NF-κB activation and the subsequent stimulation of MyD88, p38 MAPK, c-Jun N-amino-terminal kinase (JNK), and (PKCβ). The activation of these signaling pathways leads to the production of many proinflammatory cytokines, chemokines, cellular ligands, growth factors, leukocyte adhesion molecules (e.g., IL-1β, IL-6, IL-23, TNF-α, TGF-β1, MCP-1, macrophage inflammatory protein 1β (MIP-1β), granulocyte monocyte-colony stimulating factor (GM-CSF), and prostaglandin E2 [31]), ROS, nitric oxide (NO), inducible nitric oxide synthase (iNOS), and various receptors[16][20][21]. All of these molecules contribute to inflammation-related kidney injury in diabetic nephropathy.
Further immune response facilitation in diabetic nephropathy is caused by the activation of cytokine, chemokine, and angiotensin receptors that influence signaling through Janus kinase-signal transducer and activator of transcription (JAK-STAT) proteins, the mRNA levels of which correlate with the progression of diabetic nephropathy. Urine levels of MCP-1 increase in DM patients with micro- and macroalbuminuria. Moreover, MCP-1 levels correlate with the urine albumin/creatinine ratio and a decrease in renal function. In diabetic patients with macroalbuminuria, MCP-1 levels are a predictor of the progression of nephropathy. The course of the disease may also be monitored by measuring the level of other inflammation-related molecules, such as soluble TNF receptor-2 (sTNFR-2), which is associated with a decrease in the GFR. A ligand of sTNFR-2, TNF-α, and some of the other aforementioned molecules (e.g., IL-1β, IL-6, PGE2, and TGF-β1) are responsible for interstitial kidney tissue destruction through the generation of fibrotic lesions [16]. Additionally, TNF-α expression in mice was shown to be higher in a wide variety of cells by high glucose levels, AGEs, and many RAGE ligands, thereby promoting the development of obesity-induced insulin resistance[21][22]. Moreover, TGF-β1 and TNF-α decrease the expression of nephroprotective Klotho and PGC-1α proteins, which can protect kidney tissue against many harmful factors, such as ROS, and act against many of the aforementioned inflammatory, fibrotic, and apoptotic processes during the course of diabetes and diabetic nephropathy[23][24].
5. Antiinflammatory Actions of Klotho in Diabetes and Diabetic Nephropathy
Klotho expression is downregulated during inflammation. Klotho induces antiinflammatory reactions and suppresses proinflammatory NF-kB activation. Klotho inhibits TLR4 signaling and related oxidative stress. It also reduces oxidative stress by inhibiting IGF-1 signaling and NLRP3 inflammasome activation. Klotho reduces leukocyte infiltration of the kidneys, renal Injury, and fibrosis[1]. Therefore, Klotho may have therapeutic potential as an antiaging molecule and an alleviator of the effects of DM and DN by activating antiinflammatory processes, and inhibiting proinflammatory and profibrotic processes.Therefore Klotho is considered as a promising agent in immunotherapy for diabetes and diabetic nephropathy.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22020956
References
- Buendía, P.; Ramírez, R.; Aljama, P.; Carracedo, J. Klotho Prevents Translocation of NFκB. Vitam. Horm. 2016, 101, 119–150, doi:10.1016/bs.vh.2016.02.005.
- Mencke, R.; Harms, G.; Moser, J.; van Meurs, M.; Diepstra, A.; Leuvenink, H.G.; Hillebrands, J.L. Human alternative Klotho mRNA is a nonsense-mediated mRNA decay target inefficiently spliced in renal disease. JCI Insight 2017, 2, 10117294375, doi:10.1172/jci.insight.94375.
- Kim, J.H.; Xie, J.; Hwang, K.H.; Wu, Y.L.; Oliver, N.; Eom, M.; Park, K.S.; Barrezueta, N.; Kong, I.D.; Fracasso, R.P.; et al. Klotho May Ameliorate Proteinuria by Targeting TRPC6 Channels in Podocytes. J. Am. Soc. Nephrol. 2017, 28, 140–151, doi:10.1681/asn.2015080888.
- Hu, M.C.; Shi, M.; Zhang, J.; Pastor, J.; Nakatani, T.; Lanske, B.; Razzaque, M.S.; Rosenblatt, K.P.; Baum, M.G.; Kuro-o, M.; et al. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010, 24, 3438–3450, doi:10.1096/fj.10-154765.
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues—With and Without Klotho. Front. Endocrinol. 2018, 9, 189, doi:10.3389/fendo.2018.00189.
- Bian, A.; Neyra, J.A.; Zhan, M.; Hu, M.C. Klotho, stem cells, and aging. Clin. Interv. Aging 2015, 10, 1233–1243, doi:10.2147/cia.s84978.
- Quarles, L.D. Fibroblast growth factor 23 and α-Klotho co-dependent and independent functions. Curr. Opin. Nephrol. Hypertens. 2019, 28, 16–25, doi:10.1097/mnh.0000000000000467.
- Lim, S.W.; Jin, L.; Luo, K.; Jin, J.; Shin, Y.J.; Hong, S.Y.; Yang, C.W. Klotho enhances FoxO3-mediated manganese super-oxide dismutase expression by negatively regulating PI3K/AKT pathway during tacrolimus-induced oxidative stress. Cell Death Dis. 2017, 8, e2972, doi:10.1038/cddis.2017.365.
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Secreted klotho and chronic kidney disease. Adv. Exp. Med. Biol. 2012, 728, 126–157, doi:10.1007/978-1-4614-0887-1_9.
- Kim, S.S.; Song, S.H.; Kim, I.J.; Lee, E.Y.; Lee, S.M.; Chung, C.H.; Kwak, I.S.; Lee, E.K.; Kim, Y.K. Decreased plasma α-Klotho predict progression of nephropathy with type 2 diabetic patients. J. Diabetes Complicat. 2016, 30, 887–892, doi:10.1016/j.jdiacomp.2016.03.006.
- Jiang, W.; Xiao, T.; Han, W.; Xiong, J.; He, T.; Liu, Y.; Huang, Y.; Yang, K.; Bi, X.; Xu, X.; et al. Klotho inhibits PKCα/p66SHC-mediated podocyte injury in diabetic nephropathy. Mol. Cell. Endocrinol. 2019, 494, 110490, doi:10.1016/j.mce.2019.110490.
- Kim, A.J.; Ro, H.; Kim, H.; Chang, J.H.; Lee, H.H.; Chung, W.; Jung, J.Y. Klotho and S100A8/A9 as Discriminative Mark-ers between Pre-Renal and Intrinsic Acute Kidney Injury. PLoS ONE 2016, 11, e0147255, doi:10.1371/journal.pone.0147255.
- Su, H.; Lei, C.-T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405–405, doi:10.3389/fimmu.2017.00405.
- Buchanan, S.; Combet, E.; Stenvinkel, P.; Shiels, P.G. Klotho, Aging, and the Failing Kidney. Front. Endocrinol. 2020, 11, 560, doi:10.3389/fendo.2020.00560.
- Ding, S.; Xu, S.; Ma, Y.; Liu, G.; Jiang, H.; Fang, J. Modulatory mechanisms of the nlrp3 inflammasomes in diabetes. Bio-molecules 2019, 9, 850, doi:10.3390/biom9120850.
- Tesch, G.H. Diabetic nephropathy—Is this an immune disorder? Clin. Sci. 2017, 131, 2183–2199, doi:10.1042/cs20160636.
- Xia, H.; Bao, W.; Shi, S. Innate Immune Activity in Glomerular Podocytes. Front. Immunol. 2017, 8, 122, doi:10.3389/fimmu.2017.00122.
- Bhargava, R.; Tsokos, G.C. The immune podocyte. Curr. Opin. Rheumatol. 2019, 31, 167–174, doi:10.1097/bor.0000000000000578.
- Wang, L.; Wang, J.; Fang, J.; Zhou, H.; Liu, X.; Su, S.B. High glucose induces and activates Toll-like receptor 4 in endothe-lial cells of diabetic retinopathy. Diabetol. Metab. Syndr. 2015, 7, 89, doi:10.1186/s13098-015-0086-4.
- Li, Y.; Xia, W.; Zhao, F.; Wen, Z.; Zhang, A.; Huang, S.; Jia, Z.; Zhang, Y. Prostaglandins in the pathogenesis of kidney diseases. Oncotarget 2018, 9, 26586–26602, doi:10.18632/oncotarget.25005.
- Zhao, Y.; Banerjee, S.; Dey, N.; LeJeune, W.S.; Sarkar, P.S.; Brobey, R.; Rosenblatt, K.P.; Tilton, R.G.; Choudhary, S. Klotho depletion contributes to increased inflammation in kidney of the db/db mouse model of diabetes via RelA (serine)536 phosphorylation. Diabetes 2011, 60, 1907–1916, doi:10.2337/db10-1262.
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614, doi:10.1038/39335.
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gres-nigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831, doi:10.1038/ni.3790
- Carlsson, A.C.; Östgren, C.J.; Nystrom, F.H.; Länne, T.; Jennersjö, P.; Larsson, A.; Ärnlöv, J. Association of soluble tumor necrosis factor receptors 1 and 2 with nephropathy, cardiovascular events, and total mortality in type 2 diabetes. Cardio-vasc. Diabetol. 2016, 15, 40–40, doi:10.1186/s12933-016-0359-8.