A tremendous effort is currently devoted to the generation of novel hybrid materials with enhanced electronic properties for the creation of artificial photosynthetic systems. This compelling and challenging problem is well-defined from an experimental point of view, as the design of such materials relies on combining organic materials or metals with biological systems like redox-active proteins. Such hybrid systems can be used e.g. as bio-sensors, bio-fuel cells, biohybrid photoelectrochemical cells and nanosctuctured photoelectronic devices. Despite these efforts, the main bottleneck is the formation of efficient interfaces between the biological and the organic/metal counterparts for efficient electron transfer (ET). It is within this aspect that computation can make the difference and improve the current understanding of the mechanisms underneath the interface formation and the charge transfer efficiency. Yet, the systems considered are more and more complex, reaching (and often passing) the limit of current computation power.
- multiscale computation
- electron transfer
- bio-hybrid interfaces
- light harvesting protein
- metals
1. Introduction
The need to find new sources of energy production for our everyday consumption is one of the most pressing issues of our age. To overcome the dependence on fossil fuels and to assess renewable energy resources, innovative paths must be followed, to go beyond the conventional technologies that are already available. Moreover, considering the climate change problem and environmental pollution as a result of production and consumption of energy from fossil fuels, the need for green and renewable energy sources is a milestone which needs to be reached in the next two decades. If no necessary actions are taken, inevitably our future generations will be adversely affected. To counteract these problems, the European Union (EU) set a “Renewable Energy Directive” with a target of 55% reduction of greenhouse gas emission and reaching carbon neutrality by 2050 [1]. In the frame of these ambitious goals, the need of novel efficient materials is a priority for developing a competitive technology, with much effort already dedicated to bio-ethanol and other biofuels, novel photovoltaic technologies, including organic and perovskite solar cells, advanced batteries and supercapacitors, as well as electro- and photocatalytic systems for hydrogen production and a new class of fuel cells for transport.
A different approach to this problem is to take advantage and mimic what nature was able to perfect upon millions of years of evolution. Yet, one of the biggest challenges in modern science is to replicate in an artificial way the most basic processes performed by nature, such as photosynthesis. This fundamental process has been found to be a challenge to reproduce in laboratory settings using artificial, man-made systems/devices [2][3][4]. This is due to the many components needed from light-driven charge separation to the transport of charges between different photosynthetic reaction centers and to the final production of reducing equivalents, subsequently used for solar fuels synthesis. Although almost all of these steps are nowadays fully understood, mimicking this complex photosynthetic machinery in the full solar-to-fuel artificial devices that use water as the sole source of reducing equivalents is still prohibitive at an industrial scale. In particular, in the last five years research effort has been focused on the creation of an “artificial leaf” device that is able to produce clean energy from absorption of light, with some encouraging results [5][6]. Devices with external quantum efficiency of 30% has been reported [7], yet the overall efficiency is still below 1% [8][9] due to the cell design and lack of control over the light harvesting (LH) systems orientation.
2. Protein/Surface Interactions
The realization of artificial photosynthesis devices often takes place in an all-solid state device in which the protein is directly or indirectly interacting with a metal surface, to control the ET flow [5][8][9]. In addition, often the LHC is interacting with another small protein which act as shuttle of electrons (such as cytochrome), facilitating the ET process. Thus, protein adsorption on a surface is an interesting challenge for computation, as many different interactions are present, such as hydration, conformational changes and orientation of the protein to enhance the ET to/from the surface. These aspects become important when the design of novel bio-compatible applications is considered, like for biosensors [10], drug delivery systems [11] and bio-hybrid materials [12]. Experimental observations of protein absorption on a substrate have focused mainly on morphology [13], structural deformation of the protein upon adsorption [14], the effect of the surface chemistry [15] as well as kinetic of ET [16]. Yet, it is still difficult to assess the single-protein microscopic details from experimental observation, as they mainly consider thin layers or aggregate states. Computation plays once more an important role, considering MD simulations, capable to provide not only atomistic details on the adsorbed configurations, but also the time evolution of few hundreds of nanoseconds, even up to microseconds for small proteins [17] The adsorption process commonly involves the presence of a solvent (normally water) and the vast amount of molecules makes systematic studies still challenging. A common approach [18] consists of considering first an implicit solvent to determine the most energetically favorable protein orientations on the surface, followed by relaxation of the whole system including the explicit solvent molecules. Despite the reliability of this approach, it is still difficult to describe the whole absorption process from computation [19] and often this first step is skipped, with more focus oriented toward the description of the properties of the already formed protein–substrate interface, such as the intensity and direction of ET.
When immobilized on a surface, the ET occurs between the surface and redox center of the protein, giving rise to a transition from electronic to ionic charge transfer, as the electrons have to leave/enter the protein, causing the oxidation/reduction of the redox center, which is commonly observed by cyclic voltammetry measurements. This change in ionization of the protein is accompanied by changes in the environments (as it can be also charged, due to the ET process) and reduces the electrostatic barrier for subsequent steps by electrical screening [20]. If this process take places in a system in which the electrolyte is absent, or is not directly involved in the ET process, it is often termed as electron transport, and is generally considered in a solid state configuration [21]. As in this last process ions from the electrolyte are not present, charge balance is not possible and the whole process requires the electronic conduction across the whole protein to reach the electrode (or vice versa) which is defined as the electron flow through/across the protein which is in contact with the surface [22]. Computational approach to biomolecules adsorbed on surfaces can be strongly beneficial to experimentalists not only to unravel the ET mechanisms, but also to predict the behavior of biological systems with details that are not yet accessible by experimental techniques. To assess these processes at the interfaces, as for processes described in previous sections, a variety of computational methods and approaches can be considered, ranging from MD to sample the conformations of the protein adsorbed on the surface, to QM, QM/MM and ab-initio molecular dynamic (AIMD) to assess the ET and transport in both ground and excited states. For a comprehensive review on the strength and kind of different applicable methods, we refer to [23][24]. We would like to briefly remind in here how the photosynthetic devices are obtained by experiments. While the assembly is done in the presence of a solvated environment (i.e., water molecules with physiological concentration of ions), the final device is in a full-solid state, in which the solvent has been dried up. This brings additional challenges to computations, since now two different pathways can be considered to compute the adsorption and ET processes. In a first approach, in which the focus is the adsorption process, the protein and surface are immersed in a solvent environment, which is essential for the correct modeling of the process. In a second approach, in which the focus is on the DET, the protein is already considered adsorbed on the surface, and the solvent is neglected (apart from structural water molecules) to model the final experimental device.
2.1. Protein–(Self-Assembly Monolayer (SAM))–Metal Interface
MD is the method of choice when conformational studies are considered. Generally, these models consist of an idealized structure of the solid surface, which might not be representative of the actual experimental system, and simplifications on both the bond-topological structure and protein structure, protonation and interfacial solvent structure often affect the setting up of the MD simulations. Noble metals such as silver and gold are the most common metals used in experimental applications, like biosensors and bioelectronic electrodes [25][26]. The substrate is commonly considered crystalline, while in experiments often amorphous or polycrystalline surfaces are present. This latter case is especially interesting, since changing the adsorption facet of i.e., Au for a protein, can lead to strongly facet-dependent results [27]. In fact, for flat, planar Au surface, the facet of choice is the Au (111), as it is assumed to be the dominant one. While this is true for planar interfaces, more care must be payed when nanoparticles are considered, since there is still debates on which facet is the predominant one. Moreover, the presence of structural defects on the surface, as well as the presence of various functionalizations might have a strong impact on the protein conformations, and shall not be neglected when such analyses are planned from a computational side.
In addition, often self-assembly monolayer (SAM) are present in between the metal and the protein. SAM are thin films of molecules which spontaneously adsorbed in an ordered assembly over the metal (often Au). Typically, SAM are chemisorbed on the substrate through their reactive anchoring group (i.e., thiol or silate) to form a very stable assembly, and present a tail group which can be functionalized by small organic groups or even proteins. Together with the length of the SAM forming molecule itself, these characteristics determine a change in the physico-chemical properties of the metal surface and can be adjusted accordingly to the desired application. Moreover, the presence of a SAM allows for specific absorption interaction with the protein, limiting and sometime imposing a specific orientation of the biological component on the SAM, which, in turn determine the ET ability of the interface. Finally, SAMs act as electrical insulators, passivating the metal surface [28], protecting if from degradation (i.e., in case of metal oxides).
One well known system often use in computation is cytochrome c (cyt). Cyt plays an important role in a variety of artificial bio-applications like biosensors, bioelectronic devices and biofuel cells, and its adsorption on a SAM covered gold surface has been extensively studied. For these applications, the knowledge and ability of orienting the cyt on the SAM is crucial, as on it depends the ET ability of the whole interface. It has been reported that to obtain a fast ET, the protein should be oriented with the heme ring close to the SAM, in a perpendicular orientation [29] (Figure 1).
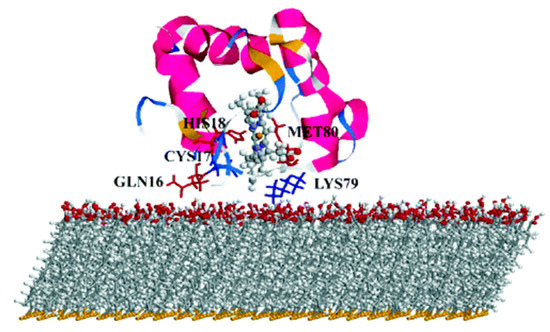
Figure 1. Simulation-predicted electron-transfer pathway of cyt on negatively charged surfaces. Reprinted with permission from [29]. Copyright (2004) American Chemical Society.
By mean of combined Monte Carlo and MD simulations, the authors of this work assessed the conformation and orientation of cyt adsorbed on a negatively charged carboxyl-terminated SAM-Au surface. Cyt could orient itself with the heme group perpendicular to the surface, forcing a specific direction of the dipole moment which is a key parameter on the determination of the final orientation. On the basis of these results, the authors indicate a possible ET pathway, from the iron of heme to the surface through a series of specific amino acids. Moreover, they warn the readers on the effect of a charged surface, which can be detrimental for the function of the protein itself.
Beside cytochrome, multiheme cytochrome (Mcyt) are a family of proteins in which two or more heme center are present, with Fe-Fe distance shorter than 1.55 nm [30][31]. Mcyt have the unique capability of long-range ET, even up to several micrometers [32], which can be applied for redox catalytic activities and electron storage [30][33]. Conversely to one heme group cyt which are widely study, the absorption on a surface and its relative ET of Mcyt is still not well understood. Mcyt can form a network of ET pathways which facilitate the total ET, and can have potential applications in i.e., solar-conversion and bioenergy [32][34]. Wei et al. [35] studied a decaheme cyt folded in four domains adsorbed on a gold substrate via thiol bonding interactions (Figure 2). The aim of their work was to assess which orientation and conformation of the adsorbed protein on gold will favor an efficient ET. Due to the complexity of the system studied, the authors devised a multiscale computational protocol in which full atomistic MD coupled with the molecular mechanics/Poison Boltzmann surface area (MM-PBSA) method are used to predict the configurations of the adsorbed protein on Au (111) surface. MM-PBSA has been shown to yield accurate prediction of the binding free energy of protein-surface interactions [36] and provides quantitative information on the driving and barrier forces of protein adsorption and docking. In the next step of their protocol, the authors perform Kinetic Monte Carlo (KMC) simulations [37] to study the ET across the protein and the hopping of electrons among the Fe atoms of the heme groups and their transfer to the surface. ET rate was quantified by using the nonadiabatic rate equation from the Marcus theory and the total ET flow assessed (Figure 2) [38].
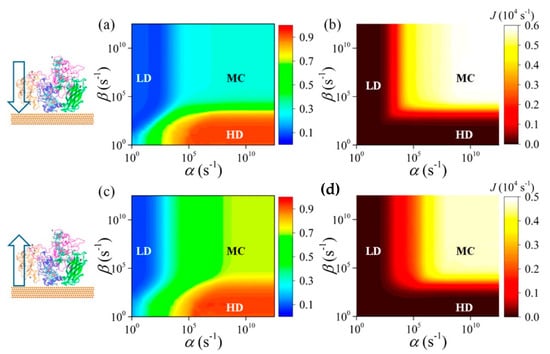
Figure 2. (a,c) Phase diagram of the time-averaged electron occupation density ⟨n⟩ for all 10 hemes in adsorbed structure as a function of the incoming (α) and outgoing (β) electron transfer (ET) rates for the transfer direction heme10-to-heme5 (from the environment to the surface through the protein) and heme5-to-heme10 (from the surface to the environment through the protein), respectively. (b,d) The corresponding phase diagrams of the net electron flux J of adsorbed protein for the transfer direction heme10-to-heme5 and heme5-to-heme10, respectively. Reprinted with permission from [35]. Copyright (2016) American Chemical Society.
As a result of this protocol, the authors were able to predict the Mcyt adsorption and ET rate when interacting with a flat gold surface. They demonstrated that the orientation of the protein, which in turn controls the ET flow, is controlled by the dehydration of the surface. Moreover, they found that this flow is more effective when the electrons move towards the interface, while is smaller in the reverse direction.
Another widely used PPC in computational studied is azurin, yet only few studies reported a computational protocol when adsorbed on a metallic surface [20][39]. Once again, MD is the method of choice to assess the conformation and orientation of the protein on the surface, as well as the retention of its folding upon adsorption. Ortega et al. [40] performed the study on an azurin (and several single amino acid mutations) adsorbed on gold surface without specific interaction, by mean of long MD simulations (up to 0.5 μs) to describe the structural changes and dynamic of azurin adsorption. The authors found that the presence of a single amino acid mutation quenches the flexibility of some region of the protein, making it stiffer. This increase in stiffness affects the adsorption dynamics on gold; the wild type adsorbs with two preferential configurations (lying-down and anchored via hydrophobic parch) thanks to the higher mobility which allows for a reorientation of the structure during the adsorption process. On the other hand, the stiffer mutants cannot easily reorient, and the final adsorption geometry is strongly dependent on the initial orientations, affecting the stability of the interface and, in turn, the ET efficiency.
2.2. Adsorption on Low Dimensional Materials
Low 1D and 2D dimensional materials are emerging as promising candidates not only as support layers due to their peculiar electronic properties, but also for active center for charge separation and storage [41]. The archetype for this materials are carbon allotropes, in particular carbon nanotubes (CNT) and single layer graphene (SLG) and due to their extremely high surface area, they are optimal candidates for the design of highly efficient biosensor applications [42]. The unique electronic characteristics of these materials is the presence of a strongly delocalized π electrons clouds which strongly favor the creation of van der Waals interactions with organic molecules and proteins.
As for the previous section, computational approaches and methods has been mainly used to describe protein/low dimensional material interaction in terms of MD simulations, to describe the adsorption process. In the last few years, rigorous approaches have been developed in order to meet the strong demands of conformational samplings [43] needed for this kind of protein/material interactions since they are often intrinsically disordered. Moreover, the size of the system pushes classical MD to its boundary, and to have meaningful results, MD should be long enough to let the protein fully adsorb and relax on the surface. To overcome these bottlenecks, Walsh et al. developed an “economical” polarizable all-atom force field to describe a graphene/water/peptide interface used in combination with replica exchange solute tempering MD (REST-MD) simulations [44]. This method is based on a replica exchange Hamiltonian-based approach that allows for efficient conformational sampling of complex interfaces. As results, the authors found that a strong binding is obtained not only for aromatic residues of the peptide, but also for residues possessing amide groups, like Asn and Gln, of a short peptide sequence. This study was performed in diluted solution for one (short) peptide chain, in order to avoid peptide-peptide interactions, which might complicate the description of the force field. In a subsequent study [45], the same authors considered the presence of multiple peptides to study their aggregation and organization on SLG/water interface, for the first time in an all-atom approach focused on advanced conformational sampling. They concluded that despite the presence of a considerable degree of peptide-peptide interactions, the interaction with the surface still resemble the one obtained from the monopeptide system, without leading to self-organization.
Moving a step forward toward complexity, Kim et al. investigated the adsorption of a peptide on SLG supported on gold as well as on multilayer graphene [46]. The result of this study was the negligible influence of the supporting layers (either gold or SLG) on the adsorption properties of short peptides. Yet, the authors did not consider the SLG/Au interactions explicitly, which might be important in the determination of the adsorption process. Challenges are still present when modeling these interfaces, such as how to consider the commensuration of the SLG/Au lattice parameters and to what point a mismatch between the two lattices is acceptable.
Graphene and its soluble derivative graphene oxide (GO) hold great promises also for the creation of biological interfaces and can be considered as materials of choice for the regulation of ET of adsorbed proteins [47][48][49], not only due to their peculiar electronic properties, but also to the high surface area. It has been reported that these materials can accommodate a vast range of redox proteins and facilitate a rapid ET through the layered materials [50]. Moreover, adsorption of cyt c over GO/reduced GO (rGO) resulted in a modulation of the protein activity due to the changes in the heme micro environment caused by the different interactions between the protein and the materials [51]. Zhao et al. [52] reported the study of cyt c adsorption over graphene and GO by mean of parallel tempered Monte Carlo (PTMC) algorithm coupled with MD simulations to assess the orientation of protein on the surface, the nature of the interactions, the subsequent conformational changes and the ET pathway. With the PTCM method, cyt c is considered in a united-residue model in which each amino acid is reduced to a site centered interaction at the carbon alpha, and the structure of the whole protein is kept rigid. The graphene/GO surface is regarded structureless and the interactions with the protein described with both vdW and electrostatic interactions. At the end of this process, optimal orientations are obtained and the most favorable were chosen as input structures for MD simulations. This protocol replaces the traditional docking procedure which is commonly performed when protein–protein interactions are present, but fails for heterogeneous interfaces as the one reported here.
From MD simulations the authors obtained that hydrophobic interactions determine the cyt-surface adsorption, with the heme group parallel to the graphene surface and almost perpendicular when adsorbed over GO (which in this study was negatively charged). The authors concluded that ET for the cyt/graphene interface is thus inhibited, while it is enhanced for the cyt/GO interface. Following recent studies showing that the ET of immobilized cyt is controlled by the interplay between tunneling probability and protein dynamics [53], the authors defined key parameters for the ET process: Redox center location, orientation of the protein on the surface and ET distance. From this study, the authors obtained that in order to favor ET tunneling (which exponentially decays with the distance) the minimal distance between the heme iron atom and the surface should be not higher than 1.3 nm. They obtain a similar distance for the protein adsorbed on GO, which should show efficient ET (Figure 3), while should be less pronounced when adsorbed on graphene, as the distance is now increased up to 1.8 nm. They rationalize these results by considering the different orientation of the heme group relative to the surface.
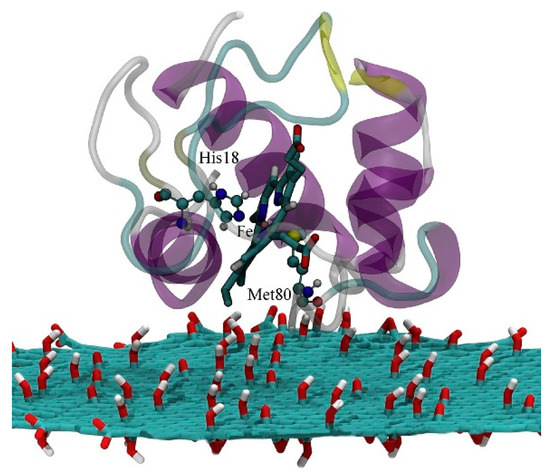
Figure 3. The electron transfer pathway of Cyt c on the graphene oxide (GO) surface. Reprinted with permission from [52]. Copyright (2018) Elsevier.
This entry is adapted from the peer-reviewed paper 10.3390/nano11020299
References
- Cabuzel, T. 2030 Climate Target Plan. Available online: https://ec.europa.eu/clima/policies/eu-climate-action/2030_ctp_en (accessed on 30 November 2020).
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473.
- Cao, R.; Lai, W.; Du, P. Catalytic Water Oxidation at Single Metal Sites. Energy Env. Sci. 2012, 5, 8134–8157.
- Blankenship, R.E.; Tiede, D.M.; Barber, J.; Brudvig, G.W.; Fleming, G.; Ghirardi, M.; Gunner, M.R.; Junge, W.; Kramer, D.M.; Melis, A.; et al. Comparing Photosynthetic and Photovoltaic Efficiencies and Recognizing the Potential for Improvement. Science 2011, 332, 805–809.
- Janna Olmos, J.D.; Kargul, J. A Quest for the Artificial Leaf. Int. J. Biochem. Cell Biol. 2015, 66, 37–44.
- Osella, S.; Kargul, J.; Izzo, M.; Trzaskowski, B. Architecture and Function of Biohybrid Solar Cell and Solar-to-Fuel Nanodevices. In Theory and Simulation in Physics for Materials Applications: Cutting-Edge Techniques in Theoretical and Computational Materials Science; Levchenko, E.V., Dappe, Y.J., Ori, G., Eds.; Springer Series in Materials Science; Springer International Publishing: Cham, Switzerland, 2020; pp. 227–274.
- Kamran, M.; Delgado, J.D.; Friebe, V.; Aartsma, T.J.; Frese, R.N. Photosynthetic Protein Complexes as Bio-Photovoltaic Building Blocks Retaining a High Internal Quantum Efficiency. Biomacromolecules 2014, 15, 2833–2838.
- Kiliszek, M.; Harputlu, E.; Szalkowski, M.; Kowalska, D.; Unlu, C.G.; Haniewicz, P.; Abram, M.; Wiwatowski, K.; Niedziółka-Jönsson, J.; Maćkowski, S.; et al. Orientation of Photosystem I on Graphene through Cytochrome C553 Leads to Improvement in Photocurrent Generation. J. Mater. Chem. A 2018, 6, 18615–18626.
- Ocakoglu, K.; Krupnik, T.; van den Bosch, B.; Harputlu, E.; Gullo, M.P.; Olmos, J.D.J.; Yildirimcan, S.; Gupta, R.K.; Yakuphanoglu, F.; Barbieri, A.; et al. Photosystem I-Based Biophotovoltaics on Nanostructured Hematite. Adv. Funct. Mater. 2014, 24, 7467–7477.
- Lasseter, T.L.; Clare, B.H.; Abbott, N.L.; Hamers, R.J. Covalently Modified Silicon and Diamond Surfaces: Resistance to Nonspecific Protein Adsorption and Optimization for Biosensing. J. Am. Chem. Soc. 2004, 126, 10220–10221.
- Basset, C.; Harder, C.; Vidaud, C.; Déjugnat, C. Design of Double Stimuli-Responsive Polyelectrolyte Microcontainers for Protein Soft Encapsulation. Biomacromolecules 2010, 11, 806–814.
- Messersmith, P.B. Multitasking in Tissues and Materials. Science 2008, 319, 1767–1768.
- Sousa, S.R.; Moradas-Ferreira, P.; Saramago, B.; Viseu Melo, L.; Barbosa, M.A. Human Serum Albumin Adsorption on TiO2 from Single Protein Solutions and from Plasma. Langmuir 2004, 20, 9745–9754.
- Wei, T.; Kaewtathip, S.; Shing, K. Buffer Effect on Protein Adsorption at Liquid/Solid Interface. J. Phys. Chem. C 2009, 113, 2053–2062.
- Su, T.J.; Green, R.J.; Wang, Y.; Murphy, E.F.; Lu, J.R.; Ivkov, R.; Satija, S.K. Adsorption of Lysozyme onto the Silicon Oxide Surface Chemically Grafted with a Monolayer of Pentadecyl-1-Ol. Langmuir 2000, 16, 4999–5007.
- Kim, D.T.; Blanch, H.W.; Radke, C.J. Direct Imaging of Lysozyme Adsorption onto Mica by Atomic Force Microscopy. Langmuir 2002, 18, 5841–5850.
- Shaw, D.E.; Maragakis, P.; Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Eastwood, M.P.; Bank, J.A.; Jumper, J.M.; Salmon, J.K.; Shan, Y.; et al. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science 2010, 330, 341–346.
- Wei, T.; Mu, S.; Nakano, A.; Shing, K. A Hybrid Multi-Loop Genetic-Algorithm/Simplex/Spatial-Grid Method for Locating the Optimum Orientation of an Adsorbed Protein on a Solid Surface. Comput. Phys. Commun. 2009, 180, 669–674.
- Wei, T.; Carignano, M.A.; Szleifer, I. Molecular Dynamics Simulation of Lysozyme Adsorption/Desorption on Hydrophobic Surfaces. J. Phys. Chem. B 2012, 116, 10189–10194.
- Bostick, C.D.; Mukhopadhyay, S.; Pecht, I.; Sheves, M.; Cahen, D.; Lederman, D. Protein Bioelectronics: A Review of What We Do and Do Not Know. Rep. Prog. Phys. 2018, 81, 026601.
- Bixon, M.; Jortner, J. Electron Transfer—from Isolated Molecules to Biomolecules. In Advances in Chemical Physics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; pp. 35–202.
- Rasor, J.P.; Voss, E. Enzyme-Catalyzed Processes in Pharmaceutical Industry. Appl. Catal. A Gen. 2001, 221, 145–158.
- Blumberger, J. Recent Advances in the Theory and Molecular Simulation of Biological Electron Transfer Reactions. Chem. Rev. 2015, 115, 11191–11238.
- Ozboyaci, M.; Kokh, D.B.; Corni, S.; Wade, R.C. Modeling and Simulation of Protein–Surface Interactions: Achievements and Challenges. Quart. Rev. Biophys. 2016, 49, e4.
- Liu, X.; Chu, P.K.; Ding, C. Surface Modification of Titanium, Titanium Alloys, and Related Materials for Biomedical Applications. Mater. Sci. Eng. R Rep. 2004, 47, 49–121.
- Alessandrini, A.; Salerno, M.; Frabboni, S.; Facci, P. Single-Metalloprotein Wet Biotransistor. Appl. Phys. Lett. 2005, 86, 133902.
- Walsh, T.R. Pathways to Structure–Property Relationships of Peptide–Materials Interfaces: Challenges in Predicting Molecular Structures. Acc. Chem. Res. 2017, 50, 1617–1624.
- Love, J.C.; Estroff, L.A.; Kriebel, J.K.; Nuzzo, R.G.; Whitesides, G.M. Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology. Chem. Rev. 2005, 105, 1103–1170.
- Zhou, J.; Zheng, J.; Jiang, S. Molecular Simulation Studies of the Orientation and Conformation of Cytochrome c Adsorbed on Self-Assembled Monolayers. J. Phys. Chem. B 2004, 108, 17418–17424.
- Bewley, K.D.; Ellis, K.E.; Firer-Sherwood, M.A.; Elliott, S.J. Multi-Heme Proteins: Nature’s Electronic Multi-Purpose Tool. Biochim. Et Biophys. Acta (Bba) Bioenerg. 2013, 1827, 938–948.
- Mowat, C.G.; Chapman, S.K. Multi-Heme Cytochromes—New Structures, New Chemistry. Dalton Trans. 2005, 21, 3381–3389.
- Breuer, M.; Rosso, K.M.; Blumberger, J.; Butt, J.N. Multi-Haem Cytochromes in Shewanella Oneidensis MR-1: Structures, Functions and Opportunities. J. R. Soc. Interface 2015, 12, 20141117.
- Bauß, A.; Koslowski, T. Storage, Transport, Release: Heme Versatility in Nitrite Reductase Electron Transfer Studied by Molecular Dynamics Simulations. Phys. Chem. Chem. Phys. 2015, 17, 4483–4491.
- Bachmeier, A.; Murphy, B.J.; Armstrong, F.A. A Multi-Heme Flavoenzyme as a Solar Conversion Catalyst. J. Am. Chem. Soc. 2014, 136, 12876–12879.
- Wei, T.; Ma, H.; Nakano, A. Decaheme Cytochrome MtrF Adsorption and Electron Transfer on Gold Surface. J. Phys. Chem. Lett. 2016, 7, 929–936.
- Paissoni, C.; Spiliotopoulos, D.; Musco, G.; Spitaleri, A. GMXPBSA 2.1: A GROMACS Tool to Perform MM/PBSA and Computational Alanine Scanning. Comput. Phys. Commun. 2015, 186, 105–107.
- Byun, H.S.; Pirbadian, S.; Nakano, A.; Shi, L.; El-Naggar, M.Y. Kinetic Monte Carlo Simulations and Molecular Conductance Measurements of the Bacterial Decaheme Cytochrome MtrF. ChemElectroChem 2014, 1, 1932–1939.
- Nakano, C.M.; Byun, H.S.; Ma, H.; Wei, T.; El-Naggar, M.Y. A Framework for Stochastic Simulations and Visualization of Biological Electron-Transfer Dynamics. Comput. Phys. Commun. 2015, 193, 1–9.
- Ruiz, M.P.; Aragonès, A.C.; Camarero, N.; Vilhena, J.G.; Ortega, M.; Zotti, L.A.; Pérez, R.; Cuevas, J.C.; Gorostiza, P.; Díez-Pérez, I. Bioengineering a Single-Protein Junction. J. Am. Chem. Soc. 2017, 139, 15337–15346.
- Ortega, M.; Vilhena, J.G.; Zotti, L.A.; Díez-Pérez, I.; Cuevas, J.C.; Pérez, R. Tuning Structure and Dynamics of Blue Copper Azurin Junctions via Single Amino-Acid Mutations. Biomolecules 2019, 9, 611.
- Scida, K.; Stege, P.W.; Haby, G.; Messina, G.A.; García, C.D. Recent Applications of Carbon-Based Nanomaterials in Analytical Chemistry: Critical Review. Anal. Chim. Acta 2011, 691, 6–17.
- Liu, Z.; Liang, X.-J. Nano-Carbons as Theranostics. Theranostics 2012, 2, 235–237.
- Walsh, T.R.; Knecht, M.R. Biomolecular Material Recognition in Two Dimensions: Peptide Binding to Graphene, h-BN, and MoS2 Nanosheets as Unique Bioconjugates. Bioconjugate Chem. 2019, 30, 2727–2750.
- Hughes, Z.E.; Walsh, T.R. What Makes a Good Graphene-Binding Peptide? Adsorption of Amino Acids and Peptides at Aqueous Graphene Interfaces. J. Mater. Chem. B 2015, 3, 3211–3221.
- Hughes, Z.E.; Walsh, T.R. Probing Nano-Patterned Peptide Self-Organisation at the Aqueous Graphene Interface. Nanoscale 2017, 10, 302–311.
- Kim, S.S.; Kuang, Z.; Ngo, Y.H.; Farmer, B.L.; Naik, R.R. Biotic–Abiotic Interactions: Factors That Influence Peptide–Graphene Interactions. Acs Appl. Mater. Interfaces 2015, 7, 20447–20453.
- Chung, C.; Kim, Y.-K.; Shin, D.; Ryoo, S.-R.; Hong, B.H.; Min, D.-H. Biomedical Applications of Graphene and Graphene Oxide. Acc. Chem. Res. 2013, 46, 2211–2224.
- Patila, M.; Pavlidis, I.V.; Diamanti, E.K.; Katapodis, P.; Gournis, D.; Stamatis, H. Enhancement of Cytochrome c Catalytic Behaviour by Affecting the Heme Environment Using Functionalized Carbon-Based Nanomaterials. Process Biochem. 2013, 48, 1010–1017.
- Shao, Q.; Wu, P.; Xu, X.; Zhang, H.; Cai, C. Insight into the Effects of Graphene Oxide Sheets on the Conformation and Activity of Glucose Oxidase: Towards Developing a Nanomaterial-Based Protein Conformation Assay. Phys. Chem. Chem. Phys. 2012, 14, 9076–9085.
- Vincent, K.A.; Li, X.; Blanford, C.F.; Belsey, N.A.; Weiner, J.H.; Armstrong, F.A. Enzymatic Catalysis on Conducting Graphite Particles. Nat. Chem. Biol. 2007, 3, 761–762.
- Yang, X.; Zhao, C.; Ju, E.; Ren, J.; Qu, X. Contrasting Modulation of Enzyme Activity Exhibited by Graphene Oxide and Reduced Graphene. Chem. Commun. 2013, 49, 8611–8613.
- Zhao, D.; Li, L.; Zhou, J. Simulation Insight into the Cytochrome c Adsorption on Graphene and Graphene Oxide Surfaces. Appl. Surf. Sci. 2018, 428, 825–834.
- Alvarez-Paggi, D.; Martín, D.F.; DeBiase, P.M.; Hildebrandt, P.; Martí, M.A.; Murgida, D.H. Molecular Basis of Coupled Protein and Electron Transfer Dynamics of Cytochrome c in Biomimetic Complexes. J. Am. Chem. Soc. 2010, 132, 5769–5778.